Reporte de caso
Emanuel Syndrome: first case reported in Mexico
Síndrome de Emanuel: primer caso reportado en México
Emanuel Syndrome: first case reported in Mexico
Investigación Clínica, vol. 59, no. 1, pp. 41-46, 2018
Universidad del Zulia
Received: 28 March 2017
Accepted: 27 October 2017
Abstract: Emanuel Syndrome is a rare genomic syndrome characterized by a supernumerary derivative 22 chromosome, acquired through an inherited chromosomal imbalance of translocation (11;22). It is characterized by severe mental retardation, facial dysmorphia, genitourinary anomalies and cardiac birth defects. We report the case of a 3-yearold girl who was born with a cleft palate, bilateral hip dysplasia and patent ductus arteriosus. The patient presented with a delay in her neurocognitive development, which prompted agenetic evaluation to be performed, demonstrating a supernumerary derivative (22)t(11;22). In the familial assessment, the mother and maternal grandmother’s karyotype revealed a balanced t(11;22). The clinical manifestations, genetic analysis and the inheritance pattern are consistent with the diagnosis of Emanuel syndrome. This is the first reported case of this syndrome in Mexico.
Keywords: der22, Emanuel syndrome, t(11;22), supernumerary chromosome, trisomy 11, trisomy 22.
Resumen: El Síndrome de Emanuel es un síndrome genómico poco común caracterizado por un cromosoma derivado supernumerario 22, adquirido a través de una herencia cromosómica desequilibrada de la translocación (11;22). Se caracteriza por retraso mental severo, dismorfia facial, anomalías genitourinarias y defectos cardíacos congénitos. Presentamos el caso de una niña de 3 años que nació con paladar hendido, displasia bilateral de cadera y conducto arterioso persistente. La paciente presentó retraso en su desarrollo neurocognitivo, por lo que se le realizó una evaluación genética, la cual demostró un derivado supernumerario (22) t(11;22). En la valoración familiar, los cariotipos de la madre y de la abuela materna revelaron una t(11;22) equilibrada. Las manifestaciones clínicas, el análisis genético y el patrón de herencia son consistentes con el diagnóstico del Síndrome de Emanuel. Este es el primer caso reportado de este síndrome en México.
Palabras clave: der22, síndrome de Emanuel, t(11;22), cromosoma supernumerario, trisomía 11, trisomía 22.
INTRODUCTION
Emanuel Syndrome (ES) is a genetic disorder caused by the presence of supernumerary derivative (der)(22) translocation(t)(11;22) due to an inherited chromosomal imbalance through 3:1 segregation during meiosis I, of a reciprocal translocation (11;22) from one of the progenitors (1). This genomic syndrome is characterized by the presence of facial dysmorphia (microcephaly, prominent forehead, abnormal auricles with ear pits or skin tags, down-slanting palpebral fissures, long philtrum, micrognathia and cleft palate) and severe intellectual disability with elayed development. It has also been described to be associated with the existence of congenital heart and genitourinary defects. Carriers of the balanced t(11;22) are clinically normal and the cytogenetic analysis demonstrates to have a supernumerary der(22). They are often not identified, until their children have the phenotypical characteristic.
CLINICAL REPORT
The patient is a 3-year-old girl who was sent to the genetics department for evaluation due to the fact that she was born with several birth defects and is currently presenting with a delayed psychomotor development. The patient was the product of the first pregnancy of a young non-consanguineous couple. She was born by vaginal delivery at 40 weeks’ gestational age after an uneventful pregnancy and presented with an Apgar score of 7 and a Silverman- Anderson score of 1. Length and weight for gestational age were within the normal range at birth: 50 cm (50th percentile) and 3150 g (50th percentile).
During the perinatal evaluation, she was found to have bilateral hip dysplasia, which was managed with the placement of a Pavlik harness for six months; and cleft palate, for which shehas undergone two reconstructive surgeries to correct this defect. Soon after birth, a patent ductus arteriosus was detected and medically corrected.
The patient achieved cephalic support at 24 months and was able to sit at 28 months. She has not been able to crawl or walk and she has not achieved urinary or fecal sphincter control.She has not developed any language skills other than making meaningless sounds. There have been multiple episodes of acute otitis and upper respiratory tract infections, which have resolved without any complications. She hasalso presented with one generalized tonic-clonic seizure in the course of her lifetime.
During the physical examination, we observed facial asymmetry, the presence of bilateral ear pits, hooded eyelids, myopia, micrognathia and upward slanting of palpebral fissures (Fig. 1). The hearing evaluation revealed mild bilateral sensorineural hearing loss.

Fig. 1.
The photograph demonstrates the facial features of the patient: facial asymmetry, bilateral ear pits, hooded eyelids, upward slanting of palpebral fissures and micrognathia.
A karyotype using G-band analysis was done and was found to have 47, XX, + der(22)t(11;22) (q23;q11) [16] (Fig. 2 -1). With respect to assessing the origin of the translocation, karyotyping of the parents was performed. The father’s cytogenetic study was normal (46, XY[16]), but the mother was found to have a balanced translocation (46, XX, t (11;22) (q23;q11) (Fig. 2 -2). Further genealogic evaluation was realized by performing a karyotype on thematernal grandmother who was found to have the translocation (46, XX, t (11;22) (q23;q11.2) [16]) as well.
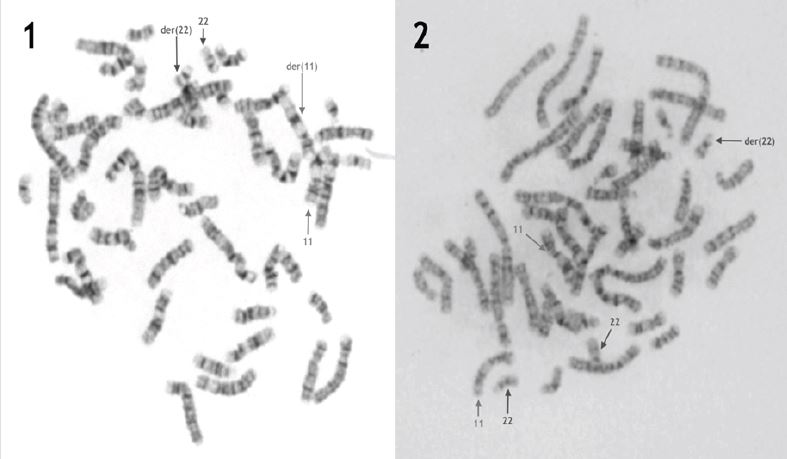
Fig. 2
1- The mother’s karyotype shows a balanced non-Robertsonian translocation between chromosome 11 and chromosome 22. 2- The patient’s karyotype shows a supernumerary derivative 22 chromosome.
We obtained a written informed consent from the parents of the patient, which gave us authorization to take photographs of the patient and use them for exhibition, publication and reproduction, with the purpose of teaching and scientific disclosure. The patient’s evaluation was done following the guidelines of the Declaration of Helsinki, amended in 2008.
DISCUSSION
Emanuel Syndrome is a clinical entity which presents with a characteristic phenotype due to an inherited chromosomal imbalance of t(11;22). The most common craniofacial characteristics include facial asymmetry, variable pre-auricular ear pits or skin tags, hooded eyelids, deepset eyes, myopia/strabismus, cleft palate, micrognathia, misaligned and crowded teeth and a hanging columella. There have also been some musculoskeletal anomalies reported such as hip dislocation, ankle instability, scoliosis, kyphosis and hypotonia. Other clinical manifestations that have been found include the presence of cardiovascular and genitourinary congenital anomalies the most common being: atrial septal defects, ventricular septal defects, patent ductus arteriosus, renal hypotrophy, micropenis and cryptorchidism. The most devastating and incapacitating manifestations are related to neurological involvement. All patients with ES present with moderate to severe mental and developmental disabilities. Gross motor and language delays are very evident in these patients; walking is achieved at age 5 and the first word at age 4, on average. It has also been reported that patients with ES, presentwith recurrent ear and upper respiratory tract infections, due to their swallowing difficulties (2). A majority of the patients present fecal and urinary incontinence, as well.
The genetic mechanism by which ES is caused, is the inheritance of one normal chromosome 22 from each parent, as well as the der(22) from the parent which is the carrier of the translocation, most commonly the mother (1). Most of the cases reported have been associated with a malsegregation 3:1 in meiosis I, during gametogenesis, in the balanced-translocation carrier. Nevertheless, there have been some case reports which imply different inheritance mechanisms such as: 2:2 segregation in the first meiotic division of the balanced translocation, followed by non-disjunction during meiosis II (3); and de novo t(11;22) in the paternal germline, with probable unbalanced adjacent 1 segregation and maternal non-disjunction of chromosome 22 during meiosis I (4).
Carriers of t(11;22) are phenotypically normal, which could be the reason it is rarely diagnosed until they present with recurrent miscarriages or give birth to a child with the unbalanced translocation. When a case of ES is diagnosed, it is important to perform a genotypic analysis in both of the parents to identify the carrier of the translocation.With regards to future pregnancies, female carriers have a higher risk (3.7%), when compared to male carriers (0.7%), of having progeny with a supernumerary der(22) t(11;22) (5).
The exact incidence is unknown, but it is a rare syndrome with around 100 reported cases. The diagnosis of Emanuel syndrome can be achieved by carefully performing a complete history and physical examination of the patient. If a chromosomal abnormality is suspected, routine cytogenetic analysis is recommended.
The supernumerary der(22) chromosome can be easily identified by routine G-bandanalysis at the 500-550 band level in individuals with this disorder. In the rare circumstance in which neither one of the parents is a balanced translocation carrier, there are commercially available FISH probes for the 22q11.2 deletion and also for the telomere of 11q which can identify the supernumerary chromosome in the karyotype as being derived from chromosomes 11 and 22. Chromosomal microarray analysis can also be employed to verify the composition of the supernumerary chromosome (6). In the case of our patient, with a highly apparent genetic alteration, it was not necessary because it was possible to confirm the diagnosis with the G-band karyotype.
This patient had the classical clinical features of Emanuel syndrome and the genetic studies performed confirmed this diagnosis. This is the first reported case of Emanuel syndrome in the Mexican population. The reproductive implications have been fully explained to the mother and cytogenetic testing has been recommended to the mother’s siblings to detect t(11;22) carriers and prevent future cases of Emanuel syndrome.
The management of Emanuel Syndrome involves a multidisciplinary approach. Because these patients present with a wide range of clinical features, they require the attention of multiple medical specialties. The treating physician should recommend a complete visual, auditory, oral, neurologic, cardiac and genitourinary evaluation, as early as possible, for thedetection and early intervention of all the congenital anomalies present (2). Early neurocognitive and psychomotor stimulation is recommended and should be offered as soon as possible to allow patients to achieve their full potential in these areas. When this patient was born, some congenital anomalies were detectedand treated but no further genetic evaluation was done, for this reason the congenital syndrome was not detected until age 3. Since the patient was not diagnosed early on, she was not able to receive the appropriate neurocognitive andpsychomotor stimulation. For this reason, physicians should stress the importance of having Emanuel Syndrome as a differential diagnosis in any patient with congenital abnormalities.
REFERENCES
1. Shaikh TH, Budarf ML, Celle L, Zackai EH, Emanuel BS. Clustered 11q23 and 22q11 breakpoints and 3:1 meiotic malsegregation in multiple unrelated t(11;22) families. Am J Hum Genet 1999; 65: 1595-1607.
2. Carter MT, St. Pierre SA, Zackai EH, Emanuel BS, Boycott KM. Phenotypic delineation of Emanuel Syndrome (supernumerary derivative 22 syndrome): Clinical features of 63 individuals. Am J Med Genet Part A 2009; 149A: 1712-1721
3. Zaki MS, Mohamed AM, Kamel AK, El- Gerzawy AMS, El-Ruby MO. EmanuelSyndrome due to unusual segregation of paternal origin. Genet Couns 2012; 23(2): 319-328.
4. Dawson AJ, Mears AJ, Chudley AE, Bech-Hansen T, McDermid H. Der(22) t(11;22) resulting from a parental de novo translocation, adjacent 1 segregation and maternal heterodisomy of chromosome 22. J Med Genet 1996; 33: 952-956.
5. Gardner RJH, Sutherland GR, Shaffer LF. Chromosome abnormalities and geneticcounseling. 4th Ed. Oxford (UK): Oxford University Press;2011
6. Emanuel BS, Zackai EH, Medne L.Emanuel Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Lebdetter N, MeffordHC, Smith RJH, and Stephens K, editors. GeneReviews® [Internet] 2007 Apr 20 [Updated 2015 Feb 5]. Available from: URL: https://www.ncbi.nlm.nih.gov/books/NBK1263/
Author notes
Corresponding author: Paloma Rivero-Moragrega Department of Genetics. Centro de Investigacion Biomedica de Occidente, Instituto Mexicano del Seguro Social. Guadalajara, Mexico. Email: paloma.rivero.m@gmail.com