INCONTINENCIA PIGMENTI: RARA GENODERMATOSIS LIGADA CON EL CROMOSOMA X. REPORTE DE UN CASO CLÍNICO
Incontinentia pigmenti: a rare X-linked genodermatosis. A case report
Incontinência pigmenti: rara genodermatose ligada com o cromossoma x. Reporte de um caso clínico
INCONTINENCIA PIGMENTI: RARA GENODERMATOSIS LIGADA CON EL CROMOSOMA X. REPORTE DE UN CASO CLÍNICO
Medicina U.P.B., vol. 35, núm. 1, pp. 52-56, 2016
Universidad Pontificia Bolivariana
Recepción: 11 Febrero 2016
Aprobación: 16 Mayo 2016
Resumen: La incontinencia pigmenti (síndrome de Bloch-Sulzberger) es una genodermatosis rara causada por una mutación en el gen NEMO localizado en el cromosoma X. Las manifestaciones cutáneas son marcadoras de esta entidad y se dan de forma temprana y cronológica. El compromiso extra cutáneo se da hasta en el 80% de los casos y las alteraciones neurológicas y oftalmológicas son marcadores del pronóstico porque tienen el potencial de producir secuelas irreversibles. Se presenta el caso de una paciente que desde el primer día de vida inició con lesiones cutáneas que, con la evolución, desarrollaron características clásicas de una incontinencia pigmenti. Se resalta el antecedente familiar asociado, el compromiso neurológico extenso y la importancia de un manejo multidisciplinario.
Palabras clave: incontinencia pigmentaria, manifestaciones cutáneas, signos y síntomas.
Abstract: Incontinentia pigmenti (Bloch-Sulzberger Syndrome) is a rare genodermatosis. The disease is X-linked and the most common molecular defect includes mutations in the NEMO gene on chromosome Xq28 in approximately 70% of patients. It is characterized by a multisystem compromise in which the skin manifestations occur in all patients. Also, the dermatological findings occur early and in chronological order. The prevalence of extra skin compromise is approximately 80% of patients. The ophthalmological and neurological manifestations are the major cause of disability in patients and may have a great impact on the quality of life. Some of damages are irreversible. We present the clinical case of a child on her first day of life. It began with skin manifestations typical of incontinentia pigmenti and extensive neurological involvement. In addition, the family history includes a sister with the disease. A multidisciplinary approach is necessary to care for patients and to detect and prevent long-term complications.
Keywords: incontinentia pigmenti, skin manifestations, signs and symptoms.
Resumo: A incontinência pigmenti (síndrome de Bloch-Sulzberger) é uma genodermatose rara causada por uma mutação no gene NEMO localizado no cromossoma X. As manifestações cutâneas são marcadoras desta entidade E se dá de forma precoce e cronológica. O compromisso extra cutâneo se dá até em 80% dos casos e as alterações neurológicas e oftalmológicas são marcadores do prognóstico porque tem o potencial de produzir sequelas irreversíveis. Se apresenta o caso de uma paciente que desde o primeiro dia de vida iniciou com lesões cutâneas que, com a evolução, desenvolveram características clássicas de uma incontinência pigmenti. Se ressalta o antecedente familiar associado, o compromisso neurológico extenso e a importância de um manejo multidisciplinar.
Palavras-chave: incontinência pigmentar, manifestações cutâneas, sinais e sintomas.
INTRODUCCIÓN
La incontinencia pigmenti es una genodermatosis rara causada por una mutación en el gen NEMO localizado en el locus Xq28, usualmente letal en hombres en el periodo prenatal. En las mujeres el fenotipo y la gravedad del cuadro clínico son extremadamente variables1,2,3,4. Hasta ahora hay alrededor de 800 casos registrados en el mundo y en el 50%-96% de ellos hay historia familiar de esta entidad3. Las manifestaciones cutáneas son sugestivas de la enfermedad y habitualmente aparecen en una secuencia cronológica caracterizada por progresión en cuatro estadios: vesicular, verrugoso, hiperpigmentado e hipopigmentado1,5. Las manifestaciones extra cutáneas se dan hasta en el 80% de los pacientes.
CASO CLÍNICO
Paciente femenina de un día de nacida, producto del cuarto embarazo de madre de 19 años con dos abortos previos, sin antecedentes patológicos. Cesárea a las 40 semanas sin complicaciones, con control prenatal normal.
Al examen físico se observan placas de color café claro con descamación fina y algunas vesículas con contenido amarillo que comprometían las cuatro extremidades (Figura 1). Se hace un diagnóstico de melanosis pustular transitoria del recién nacido y se da alta con cuidados generales de la piel y aplicación de vaselina hasta resolución de las lesiones.
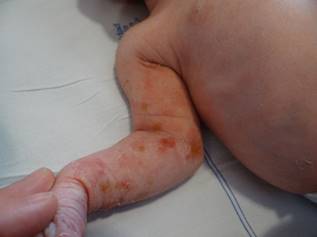
Figura 1. Lesiones en extremidades el primer día de vida.
Cuatro días después reingresa por presencia de cianosis asociada con apneas y episodios convulsivos, además, empeoramiento de las lesiones en piel y compromiso del cuero cabelludo. En el examen físico se hallan placas eritematosas infiltradas algunas con vesículas con contenido amarillo que comprometían extremidades; en miembro superior izquierdo con placas de color café claro algunas con configuración lineal y descamación en la superficie (Figura 2). En cuero cabelludo presenta placa alopécica extensa con eritema y descamación fina en la zona del vertex (Figura 3). Por el estado de la paciente se traslada a la unidad de cuidados intensivos neonatales, es evaluada por pediatra, neurología pediátrica y dermatología. La impresión diagnóstica es de un síndrome neurocutáneo en estudio y una infección por herpes virus interrogada; se ordena Aciclovir intravenoso previa toma de exámenes paraclínicos.
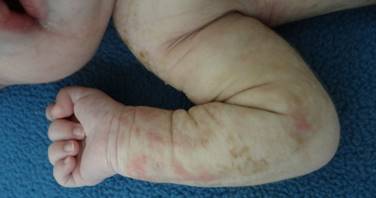
Figura 2. Lesiones en extremidades al cuarto día del nacimiento.
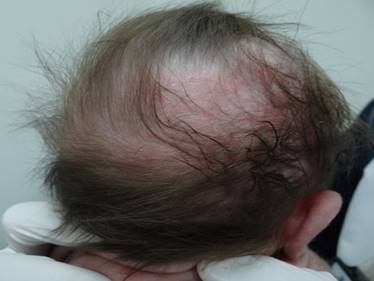
Figura 3. Lesiones en cuero cabelludo al cuarto día del nacimiento.
En los paraclínicos se reporta test de Zanck negativo, hemograma y hemocultivos normales, urocultivo normal, IgM para citomegalovirus (CMV) no reactiva, VIH negativo y líquido cefalorraquídeo (LCR) sin alteraciones con reacción en cadena de la polimerasa (PCR) para CMV y herpes virus normal.
Se realiza una resonancia magnética nuclear (RMN) cerebral que reporta áreas de encefalomalacia y gliosis que comprometen los lóbulos frontales en forma bilateral y los parieto-temporales izquierdos. Sin malformación arteriovenosa asociada.
Dermatología lleva a cabo biopsia de piel. En la evaluación histológica se observa que en la epidermis hay vesículas con eosinofilos en su interior y en la dermis, infiltrado inflamatorio linfocitario perivascular superficial: hallazgos que sugieren incontinencia pigmenti en fase vesicular (Figura 4).
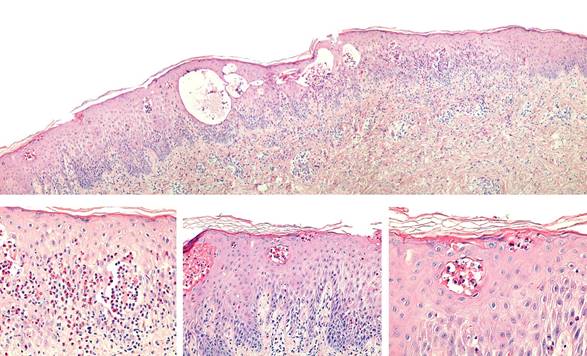
Figura 4. Microfotografías a pequeño aumento (arriba 20X), mediano (abajo izquierda y centro 100X) y gran aumento (abajo derecha 400X). Se observan algunos queratinocitos necróticos y exocitosis de eosinófilos con formación de colecciones intra epidérmicas característicos del estadio I (vesicular) de la incontinencia pigmenti.
Se re-interroga a la madre de la paciente, quien refiere antecedente de incontinencia pigmenti en su otra hija y en tía materna.
Se hace un diagnóstico final de una incontinencia pigmenti con compromiso neurológico grave, se ordena evaluación por oftalmología, que descarta, en el momento, compromiso ocular. Luego de alcanzar la estabilidad clínica de la paciente se da alta con ácido acetil salicílico, terapia anticonvulsivante, tratamiento para las lesiones de piel, tipificación genética, potenciales evocados auditivos- emisiones otoacústicas, terapia física, valoración a su hermana y control por neurología pediátrica, oftalmología y dermatología.
A los dos meses del nacimiento la paciente es llevada a urgencias por episodios convulsivos asociados con aumento progresivo de las lesiones en piel. Al examen físico hay múltiples erosiones que comprometen, principalmente, extremidades y placas gruesas de aspecto verrugoso de color amarillo-grisáceo, que predominan en los miembros inferiores (Figura 5).
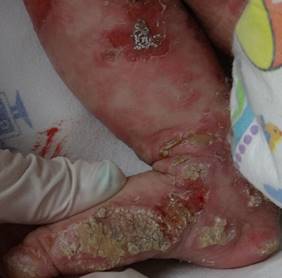
Figura 5. Lesiones en extremidades a los dos meses de vida.
DISCUSIÓN
La incontinencia pigmenti es una genodermatosis causada por una mutación en el gen NEMO localizado en el locus Xq28. Usualmente es letal en hombres en el periodo prenatal. En las mujeres el fenotipo y la severidad del cuadro son extremadamente variables. Fue descrita por Garrod en 1906 y posteriormente definida por Bloch en 1926 y Sulzberger en 1928 como Incontinentia Pigmenti, con base en los hallazgos histopatológicos característicos, aunque no patognomónicos, de las lesiones cutáneas de la tercera fase de la enfermedad1,2,3,4.
Hasta ahora hay alrededor de 800 casos registrados en el mundo y en el 50%-96% de los casos hay historia familiar de esta entidad3.
Debido a que este síndrome afecta las estructuras derivadas del neuroectodermo y mesodermo, las manifestaciones clínicas abarcan alteraciones en piel, dientes, ojos, sistema nervioso y esqueleto4.
Las manifestaciones cutáneas son indicativas de la enfermedad y habitualmente aparecen en una secuencia cronológica caracterizada por una progresión en cuatro estadios1,5:
Estadio 1. Llamado estadio inflamatorio o vesicular. Caracterizado por el desarrollo de pápulas, vesículas o pústulas en una base eritematosa distribuidas a través de las líneas de Blashko. En ocasiones esta fase se confunde con herpes simple o impétigo. Este estadio ocurre en el 90% de los pacientes y en más del 90% de estos están presentes al nacimiento o durante las primeras dos semanas de vida, y desaparece, posteriormente, a los cuatro meses de vida1,5.
Estadio 2. Estadio verrugoso. Caracterizado por placas y pápulas verrugosas lineales sobre una base eritematosa que siguen las líneas de Blashko. En general, las lesiones se desarrollan en las extremidades y el tronco. La ocurrencia de esta fase se reporta en el 70% de los casos. En muchos pacientes ocurren a las dos a seis semanas de vida y, usualmente, desaparecen a los seis meses de vida, ocasionalmente persisten en la edad adulta1,5.
Estadio 3. Estadio de hiperpigmentación. Desarrollo de lesiones lineales o circulares con una pigmentación café que puede asociarse con atrofia. Ocurre en el 90-98% de pacientes. Compromete, principalmente, el tronco, extremidades y pliegues. Estas lesiones aparecen durante el primer mes de vida y desaparecen lentamente durante la adolescencia. Sin embargo, las áreas de hiperpigmentación persisten en algunos pacientes hasta los 40 años1,5.
Estadio 4. Estadio de atrofia o hipopigmentación. Se caracteriza por lesiones atróficas, hipopigmentadas, con ausencia de folículos, principalmente localizadas en miembros inferiores. Usualmente se desarrollan durante la adolescencia, persisten en la adultez y llegan a ser permanentes. Se observan en el 30%-75% de pacientes1,5.
El cuero cabelludo es afectado en el 28%-38% de pacientes. La afectación se caracteriza por alopecia cicatricial usualmente en vertex1.
Las manifestaciones extra cutáneas se dan en el 80% de los pacientes, de estas el compromiso ocular y oftalmológico marcan el pronóstico de la enfermedad. El compromiso oftalmológico ocurre en el 35%-75% de pacientes, usualmente, se asocia con afectación neurológica. La ceguera unilateral o bilateral es reportada en el 7%-23% de los pacientes, estos pacientes también pueden presentar cataratas y estrabismo1,3.
La prevalencia de las manifestaciones neurológicas es de aproximadamente 30%. Los síntomas son variados y consisten en convulsiones, retardo psicomotor, déficit cognitivo, hemiplejia, epilepsia, encefalopatía neonatal, encefalitis, accidente cerebrovascular neonatal e infantil, entre otros. Los accidentes cerebrovasculares son la base de las manifestaciones neurológicas en el periodo neonatal y ocurren principalmente en la primera semana de vida. Los signos radiológicos encontrados son compatibles con insuficiencia vascular e incluyen leucomalacia periventricular, cambios glióticos, daño en los ganglios basales y necrosis hemorrágica difusa1,2,3,4,5.
Otras alteraciones reportadas son deformidades dentofaciales que se dan en el 60%-80% de casos y es la principal manifestación extra cutánea, alteraciones esqueléticas y el compromiso cardiaco caracterizado por insuficiencia tricuspidea e hipertensión pulmonar1,6.
Para realizar el diagnóstico de incontinencia pigmenti es fundamental el reconocimiento de las lesiones cutáneas y la búsqueda del antecedente de la enfermedad en familiar de primer grado. El estudio histopatológico es una herramienta para confirmar el diagnóstico4.
Los hallazgos histológicos son diferentes, según cada estadio. El primer estadio presenta espongiosis con eosinófilos y formación de vesículas epidérmicas. En el segundo estadio se observa acantosis, papilomatosis irregular e hiperqueratosis, con células disqueratósicas dispersas y degeneración con vacuolización de la capa basal. En lesiones del tercer estadio se encuentran melanófagos en el infiltrado inflamatorio y melanina libre en la dermis, con ausencia o disminución de la melanina en la capa basal de la epidermis. Finalmente, en el último estadio, es frecuente observar queratinocitos apoptóticos y atrofia de los folículos pilosos, con ausencia del músculo erector del pelo4.
Respecto al manejo de estos pacientes es importante que sean evaluados por genética y un control estricto por dermatología, oftalmología, pediatría y neurología. El enfoque multidisciplinario permitirá una mejor calidad de vida en estos pacientes4,5.
Se resalta de nuestro caso el antecedente familiar en la paciente, la evolución de las lesiones cutáneas similar a lo reportado en la literatura y el compromiso neurológico extenso encontrado en pocos casos; además, se destaca la importancia de un manejo multidisciplinario que incluya dermatología, oftalmología, pediatría y neurología.
Referencias
1. Poziomczyk CS, Recuero JK, Bringhenti L, Maria FDS, Campos CW, Travi GM, et al. Incontinentia pigmenti. An Bras Dermatol. 2014; 89(1): 26–36.
2. Yang Y, Guo Y, Ping Y, Zhou X-G, Li Y. Neonatal incontinentia pigmenti: Six cases and a literature review. Exp Ther Med. 2014; 8(6): 1797–1806.
3. Marques GF, Tonello CS, Sousa JMP. Incontinentia pigmenti or Bloch-Sulzberger syndrome: A rare X-linked genodermatosis. An Bras Dermatol. 2014; 89(3): 486–489.
4. Enei L, Orellana I, Vargas X, Salazar R, Paschoal F. Incontinencia pigmenti en madre e hija. Relato de caso clínico. Rev Chil Pediatr. 2011; 82(3): 225-230.
5. Welch JL, Jimenez HL, Allen SE. Pediatric rash: dermatologic manifestations of incontinentia pigmenti. J Emerg Med. 2013; 45(2): e41–3.
6. Meuwissen MEC, Mancini GMS. Neurological findings in incontinentia pigmenti; a review. Eur J Med Genet. 2012; 55(5): 323–331.
Notas de autor
luisisriba@hotmail.com
Información adicional
CONSENTIMIENTO INFORMADO: La investigación fue
aprobada por el Comité de Ética de la institución en la que se reclutó el
paciente y por las correspondientes instituciones educativas. La madre autorizó,
mediante consentimiento informado y escrito, la publicación del caso y la
reproducción de las imágenes en una revista científica.
DECLARACIÓN DE CONFLICTOS DE INTERESES: Los autores no presentan ningún conflicto de
interés. No hubo ninguna fuente de financiación.
Forma de citar este artículo: Ríos LF, Vásquez E,
Mejía MA, García MT, Restrepo R. Incontinencia pigmenti: rara genodermatosis
ligada con el cromosoma X. Reporte de un caso clínico. Med U.P.B. 2016;35(1):52-56.