Reporte
Recepción: 10 Febrero 2022
Aprobación: 02 Agosto 2022
DOI: https://doi.org/10.11144/Javeriana.umed63-4.adam
Resumen: Introducción: La displasia geleofísica es un trastorno heterogéneo del tejido conectivo, caracterizado por presentar baja talla, manos y pies pequeños, y limitación para los movimientos articulares, con heterogeneidad de locus y herencia autosómica recesiva. Objetivo: Describir los hallazgos clínicos y radiológicos de dos pacientes con displasia geleofísica tipo 1. Casos clínicos: Recién nacido a término, con antecedente de polihidramnios, pequeño para la edad gestacional. A las 20 horas de vida presentaba limitación para los movimientos articulares, con dedos de las manos y los pies cortos, de piel gruesa y áspera. Con cuatro meses de edad, los exámenes de laboratorio reportaron: disminución de la hormona de crecimiento 0,90 ng/ml (1-9 ng/ml) y el factor de crecimiento insulinoide tipo 1 < 40 ng/ml (49-327 ng/ml) con persistencia de baja talla, por debajo del percentil 3. El estudio molecular reportó una variante que se encuentra en heterocigosis compuesta c.[215G>A] + [340G>A] en el gen ADAMTSL2, para displasia geleofísica tipo 1. El segundo caso se trata de un recién nacido de 72 horas de vida, medio hermano del primer caso, quien presentó hallazgos clínicos coincidentes con el primero, además de válvula bicúspide aórtica; ambos casos del mismo padre con diferente pareja. Conclusión: Los pacientes con displasia geleofísica tipo 1 constituyen casos de interés por tratarse de un diagnóstico poco frecuente en la edad pediátrica y la sospecha clínica puede generarse a partir de los hallazgos clínicos cardinales descritos, corroborados con el estudio molecular y complementado con la confección e interpretación de la genealogía familiar.
Palabras clave: Braquidactilia, heterogeneidad genética, mutación missense, polihidramnios.
Abstract: Introduction: Geleophysic dysplasia is a heterogeneous disorder of connective tissue, characterized by short stature, small hands and feet, limited joint movements. With locus heterogeneity and autosomal recessive inheritance. Objective: To describe the clinical and radiological findings of two patients with type 1 geleophysic dysplasia, discussion of differential diagnoses, natural history and molecular findings. Clinical cases: Term newborn, with a history of polyhydramnios, small for gestational age, was assess at 20 hours of life, who presented limitated of joint movements, with short fingers and toes, thick and rough skin. At four months of age, laboratory tests reported: decreased growth hormone 0.90 ng / ml (1-9 ng / ml) and insulin-like growth factor type 1 <40 ng / ml (49-327 ng / ml) with persistence of short stature, below the 3rd percentile. The molecular study reported a variant that was compound heterozygous c.[215G>A] + [340G>A] in the gene ADAMTSL2, for type 1 geleophysic dysplasia. The second case is a 72-hour-old newborn, half-brother of the first case, who presented clinical findings that consistent with the first, except that he also presented a bicuspid aortic valve; both cases of the same father with a different partner. Conclusion: Patients with type 1 geleophysical dysplasia constitute interes cases in the cause of rare diagnosis in pediatric age and clinical suspicion, It can be generated from the describe cardinal clinical findings, corroborated with the molecular study and complemented with the preparation and interpretation of family genealogy.
Keywords: Brachydactyly, genetic heterogeneity, mutation, missense, polyhydramnios.
Introducción
La displasia geleofísica (GPHYSD, por sus siglas en inglés) es un trastorno heterogéneo del tejido conectivo (1) que corresponde al grupo de displasias acromélicas (2), caracterizada por presentar talla baja, manos y pies pequeños, enfermedad cardiaca valvular, hepatomegalia, limitación para los movimientos articulares, piel gruesa y áspera, con inteligencia normal y dismorfismo facial leve (3).
Tiene una prevalencia desconocida, pues solo se han descrito 55 casos en el mundo (4). En Ecuador, hasta la fecha, no existen casos reportados. La GPHYSD presenta heterogeneidad genética no alélica o de locus, que son las variantes patogénicas identificadas en los genes ADAMTSL2 OMIM #231050 (tipo 1, autosómico recesivo), FBN1 OMIM #614185 (tipo 2, autosómico dominante) y en LTBP3 OMIM #617809 (tipo 3, autosómico dominante) (5,6).
Inicialmente, la GPHYSD fue considerada un trastorno de depósito lisosomal (7); más tarde, se reconoció como una de las causas las mutaciones en el gen ADAMTLS2 (A Disintegrin And Metalloproteinase with Thrombospondin repeats-like 2), localizado en el cromosoma 9q34.2. El gen tiene descritas 26 mutaciones, de las cuales 22 son con cambio de sentido (missense), dos deleciones y dos mutaciones de splicing (8,9).
La gran familia ADAMTS contiene 19 proteasas a la cual pertenece ADAMTSL2, de la cuales como mínimo 5 son proteínas ADAMTS-like. La subfamilia ADAMTS-like contiene proteínas homólogas para el dominio no ciliar (ancillary domain), es decir, con pérdida del dominio proteasa, consecuentemente con la pérdida de la actividad catalítica. Las proteínas ADAMTSL-1 y ADAMTSL-3 están estrechamente relacionadas con las glicoproteínas secretadas, donde ADAMTSL-2 tiene una estructura diferente. ADAMTSL2 presenta 18 exones que codifican para un producto proteico de 951 aminoácidos, constituido por un péptido señal, seguido por la repetición trombospondina tipo I (TSR), una región rica en cisteína, otra región espaciadora, una región rica en N-glican, seis TSR adicionales y, finalmente, una región PLAC (protease and lacunin module) (10,11,12). Isogai et al. (13) encontraron que la proteína LTBP1 interviene en el almacenamiento del TGFβ (transforming growth factor-β) en la matriz extracelular. El TGFβ regula la proliferación, migración, diferenciación y sobrevida celular. En los pacientes con GPHYSD, la interacción deficiente entre ADMSTSL2 y LTPB1 altera la producción, la activación del TGFβ y, posiblemente, la conformación de las microfibrillas.
Las displasias acromélicas son difíciles de identificar, incluso en manos expertas. A esto se suma el hecho de que las enfermedades genéticas son poco frecuentes. El objetivo de la investigación consistió en describir los hallazgos clínicos y radiológicos de dos pacientes con displasia geleofísica tipo 1, así como discutir los diagnósticos diferenciales, su historia natural y los hallazgos moleculares.
Caso clínico 1
El caso corresponde a un recién nacido (IV-8, según la genealogía) de 20 horas de vida, cuyo caso fue interconsultado al servicio de genética, por talla corta, dismorfias faciales y alteraciones esqueléticas. Entre sus antecedentes contaba: madre de 27 años de edad y padre de 29 años, mestizos, no consanguíneos, sin historia familiar relevante. El embarazo registró polihidramnios (Índice de Líquido Amniótico: mayor de 5) en el tercer trimestre de gestación; además, con una longitud de fémur con 56 mm y húmero de 51 mm, equivalentes para una edad de 32 semanas, en una gestación de 36 semanas. La madre no tenía antecedentes de consumo de alcohol o tabaco durante el embarazo. El estudio de TORCH (toxoplasma, rubéola, citomegalovirus y herpes virus) fue negativo.
El recién nacido era producto de un tercer embarazo de 38,2 semanas, según la fecha de la última menstruación. Nació a través de cesárea por pelviano, con líquido amniótico claro, con antropometría que correspondió a un recién nacido a término pequeño para la edad gestacional, según las curvas de crecimiento de la Organización Mundial de Salud (OMS), por debajo del percentil 3, con peso de 2050 g, talla de 42 cm, perímetro cefálico de 31 cm y Apgar de 8 al minuto y a los 5 minutos.
En el examen físico segmentario, se observó en su cráneo una fontanela anterior normotensa, cabello ondulado, de color negro. Facie: redonda, con mejillas rellenas, frente amplia, cejas escasas, prominente premaxilar, micrognatia; pabellones auriculares con lóbulos hipoplásicos; nariz pequeña con dorso nasal amplio, con punta nasal ancha, filtro nasal largo; boca con apariencia entreabierta, arco de cupido bien definido, con labio superior fino (figura 3c).
En el tórax se auscultaron ruidos cardiacos rítmicos con ausencia de soplos. El abdomen no evidenció visceromegalias. El recién nacido tenía una limitación generalizada para los movimientos articulares, con predominio en las extremidades superiores; así como limitación para los movimientos articulares con flexión en los codos (figura 1b). Las extremidades inferiores evidenciaron una flexión moderada de las caderas, con importante resistencia para la extensión de las rodillas, sin crepitación a la palpación, con dedos de las manos y los pies cortos. La piel estaba gruesa y áspera.
Las radiografías de las extremidades superiores e inferiores mostraron huesos largos tubulares y cortos; manos y pies con epífisis de las falanges proximales con forma de cono (figura 1a). Una vez obtenidos los resultados de los exámenes iniciales y las valoraciones médicas, el paciente fue enviado a casa en condición estable.
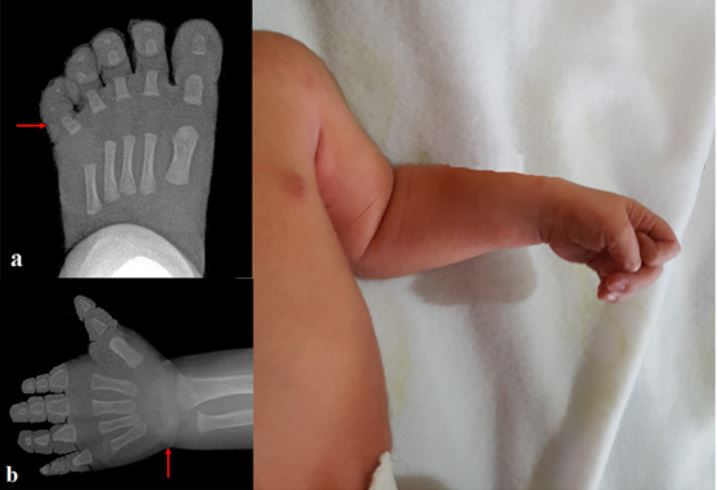
Figura 1.
a) Radiografía de pie izquierdo, que evidencia falanges proximales con epífisis en forma de cono y brazo izquierdo con dedos cortos y leve flexión de codo al nacimiento. b) Radiografía de mano izquierda con ocho meses de edad, que evidencia retraso en la edad ósea, que corresponde para una edad de tres meses.
Al control, con cuatro meses de edad, los exámenes de laboratorio reportaron: disminución de la hormona de crecimiento 0,90 ng/ml (1-9 ng/ml) y el factor de crecimiento insulinoide tipo 1 < 40 ng/ml (49-327 ng/ml) con talla de 57 cm, por debajo del percentil 3, según las curvas de crecimiento de la OMS. Considerando los hallazgos clínicos y radiológicos, se procedió a tomar una muestra de sangre periférica para realizarle un estudio molecular al paciente (IV-8), la hermana (IV-6) y su madre (III-10).
El estudio molecular se llevó a cabo por secuenciación de nueva generación utilizando el panel TruSight Inherited Disease con 2,25 Mb de contenido genómico, con 8801 exones target y 552 genes para diferentes enfermedades hereditarias. Se siguió el protocolo publicado por el fabricante y posteriormente fue corrido en el equipo MiSeq, y así se identificaron 8 variantes: 3 posiblemente patogénicas, 4 variantes de significado incierto y una variante patogénica que se encuentra en heterocigosis compuesta NM_014694.4, c.340G>A (p.E114K) y c.215G>A (p.R72Q) del gen ADAMTSL2, en el probando; mientras tanto, la madre (III-10) y hermana (IV-6) fueron heterocigotas simple (figura 2).
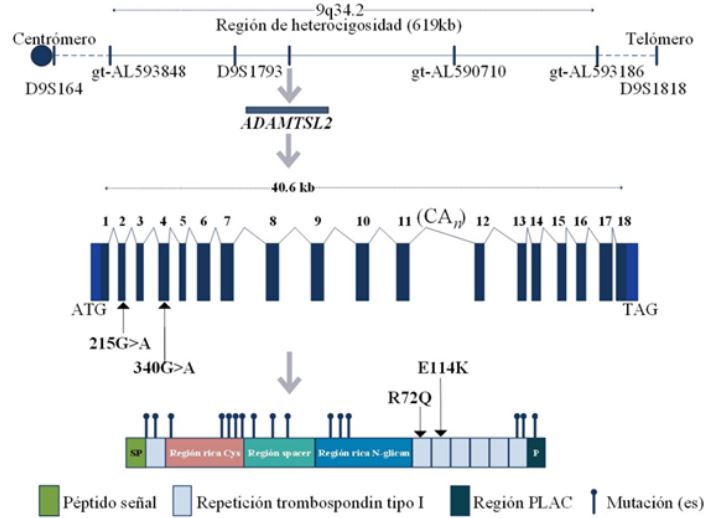
Figura 2.
Estructura, exones e intrones de ADAMTSL2 con la variante de heterocigoto compuesto c.[215G>A]+[340G>A].
Con ocho meses de edad, la evaluación radiográfica de seguimiento en la consulta externa evidenció retraso en la edad ósea, misma que correspondió aproximadamente para una edad de tres meses (figura 1b), con talla de 64 cm, persistencia entre el percentil 3 y debajo del percentil 10, según las curvas de crecimiento de la OMS. Con persistencia de la limitación generalizada para los movimientos articulares y retraso en el desarrollo motor fino, posiblemente por la falta de movilidad interfalángica. No ha presentado infecciones recurrentes respiratorias y de oído.
Caso clínico 2
Este otro caso corresponde a un recién nacido (IV-5) de 72 horas de vida, medio hermano del primer caso clínico, quien fue interconsultado al servicio de genética, con los antecedentes, madre de 31 años y padre con 29 años, mestizos, no consanguíneos. El embarazo registró cuatro ecografías normales. La madre sin antecedente de consumo de alcohol o tabaco durante el embarazo; el estudio de TORCH fue negativo.
El recién nacido era producto de un quinto embarazo. Nació por parto eutócico de 39 semanas, según la fecha de la última menstruación, con una antropometría que correspondió a un recién nacido a término pequeño para la edad gestacional, según las curvas de crecimiento de la OMS, entre el percentil 3 y debajo del percentil 10, con un peso de 2489 g, una talla de 47 cm, un perímetro cefálico de 33 cm, un Apgar de 8 al minuto y de 9 a los 5 minutos.
En el examen físico segmentario se observó que el cráneo del recién nacido tenía una fontanela posterior puntiforme, cabello liso, de color negro, con patrón de implantación variable. Facie: redonda, mejillas rellenas, frente amplia con leve hirsutismo, pestañas poco pobladas, cejas escasas, prominente premaxilar, micrognatia; pabellones auriculares; nariz pequeña con dorso nasal amplio, narinas antevertidas, filtrum nasal largo y superficial; boca con apariencia entreabierta, arco de cupido bien definido, con labio superior fino (figura 3a).
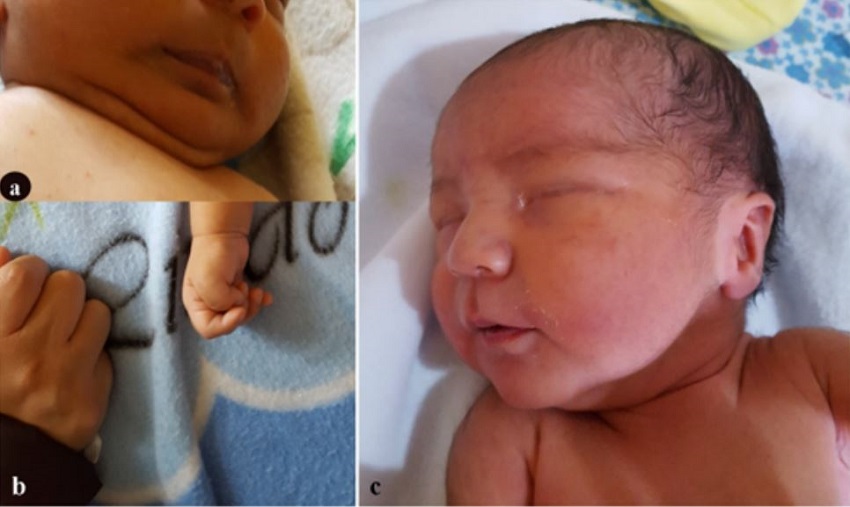
Figura 3.
Vista de tercio inferior facial, con expresión “feliz” y relación de tamaño de la mano, entre la madre y el paciente IV-5 al nacimiento (a y b, respectivamente). Vista anterolateral de macizo facial del paciente IV-8 al nacimiento (c).
En el tórax del recién nacido se evidenció un soplo sistólico, grado II, en el quinto espacio intercostal izquierdo, sin irradiación al hueco supraesternal. El abdomen no evidenció visceromegalias. Las extremidades superiores presentaron limitación para los movimientos articulares con flexión de los codos. Las extremidades inferiores evidenciaron flexión de las caderas con resistencia a la extensión en las rodillas, sin crepitación a la palpación; con dedos de las manos y pies cortos (figura 3b).
En el nacimiento, las radiografías de las extremidades superiores e inferiores mostraron huesos largos tubulares y cortos; manos y pies con falanges proximal ancha y epífisis con forma de cono. El examen cardiológico, que consistió en la valoración estructural a través de ecocardiograma, reportó válvula bicúspide aórtica, estenosis aórtica con leve dilatación postestenótica, con gradiente sistólico entre el ventrículo izquierdo y la aorta de 40 mmHg; sin aparente afectación de ventrículo izquierdo y válvula mitral con ausencia de estenosis o regurgitación.
Los hallazgos clínicos y radiológicos llaman la atención, puesto que fueron bastante coincidentes con el primer caso clínico. Cuando se confeccionó la genealogía familiar del paciente (IV-5), nos sorprendió identificar otro hijo del mismo padre (III-9), pero con otra pareja (III-8) de la misma localidad (figura 4).
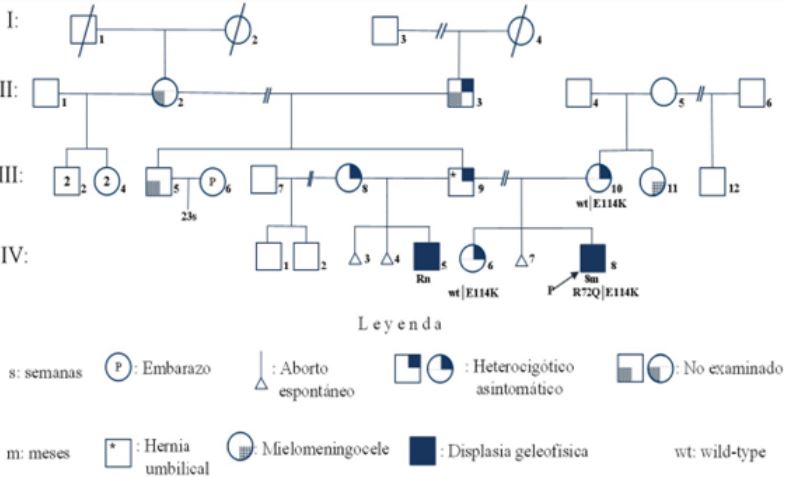
Figura 4.
Genealogía de la familia con displasia geleofísica
Al interpretar la genealogía de la familia con GPHYSD, se definió que los individuos (II-3, III-8, III-9), probablemente, eran heterocigóticos asintomáticos; mientras que III-10 y IV-6 fueron heterocigóticas, con patrón de herencia probable autosómico recesivo. En el examen físico de los individuos III-8, III-9 y III-10 se observó ausencia de talla baja, braquidactilia, marcha en puntillas y dismorfismo facial. El paciente III-9 había tenido como antecedente una hernia umbilical dos años atrás. Las radiografías de las extremidades superiores e inferiores fueron normales (no mostradas). III-8 y III-10 tuvieron en común el antecedente de abortos con la misma pareja (III-9). Ante estas circunstancias, se ofrece a III-8 y III-9 la realización de un estudio molecular para ellos mismos y para IV-5, pero no brindan su consentimiento. Con la pareja (III-8 y III-9), al encontrarse en edad reproductiva, se planificaron sesiones de asesoramiento genético.
Discusión
La GPHYSD1 tiene un patrón de herencia probable autosómico recesivo, con características faciales que incluye la facie “feliz” (del griego geleos: feliz y physis: natural), nariz pequeña con narinas antevertidas, filtrum nasal largo y labio superior fino (14), hallazgos clínicos similares con los descritos en los pacientes IV-5 y IV-8 de la familia estudiada. Entre los hallazgos radiográficos se incluyen los huesos largos tubulares; la epífisis de falanges proximal en forma de cono (15,16), que fue evidenciado en nuestros casos, y el retraso en la edad ósea en el paciente IV-8, quien con ocho meses de edad cronológica presentaba una edad ósea que correspondía a un niño de tres meses.
El fenotipo del paciente (IV-8) en la familia estudiada es el resultado de la combinación de dos alelos mutados; sin embargo, no se podría descartar la probabilidad de que el paciente IV-5, al no presentar un fenotipo diferente, sea portador de una mutación en un alelo y que el otro alelo tenga otra mutación (heterocigoto compuesto), como mecanismo patológico molecular que explique la heterogeneidad genética alélica (17).
Los pacientes IV-5 y IV-8 de la familia estudiada presentaron dismorfismo facial, coincidente con los hallazgos descritos por Allali et al. (18), quienes concluyeron la presencia de heterogeneidad genética para GPHYSD1. El estudio molecular del paciente IV-8 informó la presencia de una mutación en heterocigosis compuesto para ADAMTSL2, presente solo en el 42% de los casos diagnosticados con GPHYSD1, y de estos el 50% procede de padres no consanguíneos.
En un estudio de Allali et al. (18), con respecto a los antecedentes prenatales, se evidenció la presencia de polihidramnios y restricción de crecimiento intrauterino en el 91% y en el 76%, respectivamente, de los casos diagnosticados con GPHYSD1. Tales hallazgos coinciden con los presentados por el paciente IV-5, quien tenía comprometida la talla desde el nacimiento, por debajo del percentil 3. El paciente IV-8, a pesar de que presentó disociación entre la antropometría fetal y la edad gestacional, no fue posible precisar el diagnóstico en primera instancia en la etapa prenatal.
En el planteamiento del diagnóstico diferencial (tabla 1) deberían considerarse síndromes genéticos con patrón de herencia monogénico, que ante su alta similitud fenotípica dificultan el diagnóstico preciso (19,20,21,22).
Diagnóstico diferencial de displasia geleofísica
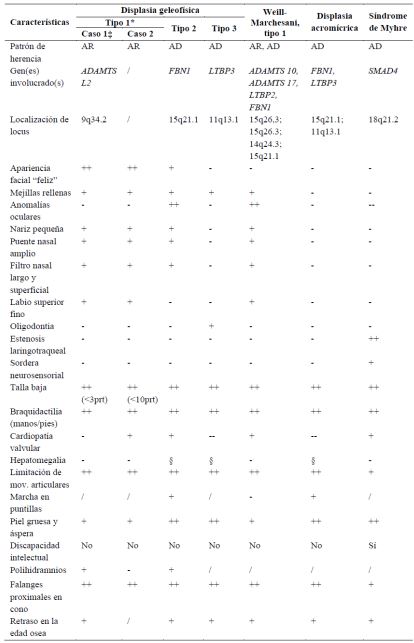
* Descripción de los casos; ‡ con estudio molecular; / dato no disponible; ++/-- de mayor importancia; § presente solo en casos severos; prt: percentil.
En la genealogía de la familia con GPHYSD, la madre (II-2), quien tuvo cuatro hijos, de los cuales dos no estaban afectados con la segunda pareja (II-1), podría sugerir que II-2 no es portadora del alelo mutado; por tanto, es probable que III-9 sea portador de un alelo mutado heredado de su padre (II-3). Las pacientes III-8 y III-10 en la familia estudiada presentaron como antecedente común abortos en el primer trimestre de embarazo con la misma pareja, lo cual, posiblemente, se deba a alteraciones en el genoma. Sin embargo, queda pendiente para futuras investigaciones.
Al interpretar la genealogía de la familia y evidenciar la ausencia de consanguinidad como elemento que modifique el equilibrio de la Ley de Hardy Weinberg (23), es poco probable que exista la unión entre dos individuos portadores de un alelo mutado. La pareja III-8 y III-9, así como III-10, al proceder de una misma población pequeña, portadores de una mutación en al menos un alelo relativamente raro, se interpreta como la presencia de un efecto fundador, donde la frecuencia de un alelo se perpetúa y supera a la población de origen.
La confección de la genealogía familiar permite definir el patrón de herencia probable en la GPHYSD en el momento de asesorar a la pareja (III-8 y III-9), puesto que la probabilidad de riesgo varía, dependiendo del patrón de herencia. Probablemente, por ser portadores, al recibir asesoramiento genético, si ellos están interesados en conocer las posibilidades que tienen de tener otro hijo o hija afectado, la probabilidad se mantiene en el 25% (riesgo alto, mayor al 15%) en cada embarazo (24) de que el niño o niña herede los dos alelos mutados.
En este estudio fue evidente la presencia de mutaciones con cambio de sentido (missense), debido a que la sustitución de un único nucleótido c.[215G>A]+[340G>A] en la secuencia del gen ADAMTSL2 altera el código en el triplete de bases; por tanto, causa evidentemente la sustitución de un aminoácido por otro p.[R72Q]+[E114K] en la región de seis TSR del producto proteico, que interactúa deficientemente con LTPB1, que a posterior altera la producción, activación del TGFβ (13).
Conclusiones
Los pacientes con GPHYSD tipo 1 constituyen casos de interés, por tratarse de un diagnóstico poco frecuente en la edad pediátrica y porque la sospecha clínica puede generarse a partir de un buen examen clínico, complementado con la confección e interpretación de la genealogía familiar en los trastornos con patrón de herencia mendeliano, que consecuentemente permitiría ofrecer el asesoramiento genético correspondiente. Los hallazgos clínicos descritos contribuyen a una alta sospecha diagnóstica que será comprobada con el estudio molecular pertinente.
Responsabilidades éticas
Protección de personas y animales. Los autores declaran que los procedimientos seguidos se conformaron a las normas éticas del Comité de Experimentación Humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos del centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores han obtenido el consentimiento informado de los pacientes o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de intereses
Los autores declaran no tener conflictos de intereses.
Referencias
Geleophysic dysplasia [database on internet]. Online Mendelian Inheritance in Man. [Citado 2021 dic 2]. Disponible en: https://www.ncbi.nlm.nih.gov/omim.
Bonafe L, Cormier-Daire V, Hall C, Lachman R, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet. 2015;Part A 167:2869-92.
Spranger J, Gilbert EF, Arya S, Hoganson GMI, Opitz JM. Geleophysic dysplasia. J Med Genet 1984;19:487-99.
García-Ortiz L, Gutiérrez-Salinas J, Chima MC, Romero RL, et al. Geleophysic dysplasia: a novel in-frame deletion of a Tandem repeat in the ADAMTSL2 gene. Am J Med Genet. 2015;Part A 167:1949-51.
Hasegawa K, Numakura C, Tanaka H, Furujo M, et al. Three cases of Japanese acromicric/geleophysic dysplasia with FBN1 mutations: a comparison of clinical and radiological features. J Pediatr Endocrinol Metab. 2016;1:1-5.
McInerney-Leo AM, Le Goff C, Leo PJ, Kenna TJ, et al. Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. J Med Genet. 2016;53:457-64.
Spranger J, Gilbert E, Tuffli G, Rossiter F, et al. Geleophysic dwarfism--a ‘focal’ mucopolysaccharidosis? Lancet 1971;298:97-8.
Le Goff C, Mahaut C, Wang LW, Allali S, et al. Mutations in the TGFb binding-protein-like Domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Gent. 2011;89:7-14.
Li D, Dong H, Zheng H, Song J, Song J, Li X, Jin Y, Liu Y, Yang Y. A chinese boy with geleophysic dysplasia caused by compound heterozygous mutations in ADAMTSL2. Eur J Med Genet. 2017;60(12):685-9. https://doi.org/10.1016/j.ejmg.2017.09.003
Ben-Salem S, Hertecant J, Al-Shamsi AM, Ali BR, Ali BR, Al-Gazali L. Novel mutations in ADAMTSL2 gene underlying geleophysic dysplasia in families from the United Arab Emirates. A Clin Molec Teratol. 2013;97:764-9. https://doi.org/10.1002/bdra.23170
Le Goff C, Morice-Picard F, Dagoneau N, Wang LW, Perrot C, Crow YJ, et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-b bioavailability regulation. Nature Genet. 2008;40(9):1119-23. https://doi.org/10.1038/ng.199
Hubmacher D, Apte S. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015;47:34-43.
Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril- associated protein. J Biol Chem 2003;278:2750-57. https://doi.org/10.1074/jbc.M209256200
Mackenroth L, Rump A, Lorenz P, Schröck E, Tzschach A. Novel ADAMTSL2-mutations in a patient with geleophysic dysplasia type I. Clin Dysmorphol. 2016;25(3):106-09. https://doi.org/10.1097/MCD.0000000000000124
Shohat M, Gruber HE, Pagon RA, Witcoff LJ, Lachman R, Ferry D, et al. Geleophysic dysplasia: a storage disorder affecting the skin, bone, liver, heart, and trachea. J Pediat. 1990;117(2 pt 1):227-32. https://doi.org/10.1016/s0022-3476(05)80534-7
Jin HS, Song Ho, Cho SY, Ki Ch-S, Yang SH, Kim OH, et al. Acromicric dysplasia caused by a novel heterozygous mutation of FBN1 and effects of growth hormone treatment. Ann Lab Med. 2017;37(1):92-4. https://doi.org/10.3343/alm.2017.37.1.92
Strachan T. Genes in pedigrees and populations. En: Strachan T, Read A, editores. Human molecular genetics. 4.ª ed. New York: Garland Science; 2011. p. 61-72.
Allali S, Le Goff C, Pressac-Diebold I, Pfennig G, Mahaut C, Dagoneau N, et al. Molecular screening of ADAMTSL2 gene in 33 patients reveals the genetic heterogeneity of geleophysic dysplasia. J Med Genet. 2011;48(6):417-21. https://doi.org/10.1136/jmg.2010.087544
Piccolo P, Sabatino V, Mithbaokar P, Polishchuk E, Hicks J, Polishchuk R, et al. Skin fibroblasts of patients with geleophysic dysplasia due to FBN1 mutations have lysosomal inclusions and losartan improves their microfibril deposition defect. Mol Genet Genomic Med. 2019;7(9):e844. https://doi.org/10.1002/mgg3.844
Hubmachera D, Tayea N, Balic Z, Thackerb S, Adamsc S, Birk D, et al. Limb- and tendon-specific Adamtsl2 deletion identifies a role for ADAMTSL2 in tendon growth in a mouse model for geleophysic dysplasia. Matrix Biol. 2019;89:38-53.
Marzin P, Cormier-Daire V, Tsilou E. Weill-Marchesani syndrome [internet]. Seattle (WA): University of Washington; 2007 [actualizado 2007 nov 1 2020 dic 10; citado 2021 dic 27]. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1114/
Starr L, Lindor N, Lin A. Myhre syndrome [internet]. Seattle (WA): University of Washington; 2017 Apr 13 [updated 2017 Apr 13; citado 2021 nov]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK425723/.
Lardoeyt R. Aplicación de la ley de Hardy Weinberg en las investigaciones científicas. En: Lardoeyt R, editor. Fundamentos de genética médica poblacional. La Habana: ECIMED; 2016. p. 31-52.
Rojas I. Prevención de enfermedades genéticas y asesoramiento genético. En: Lantigua A, editor. Introducción a la genética médica. La Habana: ECIMED; 2011. p. 342-72.
Notas
Notas de autor
a Autor de correspondencia: victor_hg7@hotmail.com
