Carátula del artículo
Artículos de Revisión de Tema
Consideraciones clínicas de la abetalipoproteinemia: revisión de la literatura
Clinical considerations of abetalipoproteinemia: literature review
 Ivan David Lozada Martínez ivandavidloma@gmail.com
Ivan David Lozada Martínez ivandavidloma@gmail.com
Universidad de Cartagena, Colombia
María Paz Bolaño Romero mbolanor1@unicartagena.edu.co
Universidad de Cartagena, Colombia
Andrea Carolina Díaz González adiazg@unicartagena.edu.co
Universidad de Cartagena,, Colombia
Amileth Suarez Causado asuarezc1@unicartagena.edu.co
Universidad de Cartagena, Colombia
Archivos de Medicina (Col), vol. 20, núm. 2, pp. 461-471, 2020
Universidad de Manizales
Recepción: 24 Noviembre 2019
Corregido: 08 Abril 2020
Aprobación: 30 Abril 2020
Introducción
Se ha descrito que la abetalipoproteinemia (ABL) es un trastorno muy poco frecuente, caracterizado por niveles bajos o ausentes de colesterol plasmático, lipoproteínas de baja densidad (LDL) y lipoproteínas de muy baja densidad (VLDL), pero no debe confundirse con una deficiencia de beta-lipoproteínas. El cuadro de presentación se basa principalmente en malabsorción de grasas, degeneración espinocerebelosa, acantocitosis y retinitis pigmentosa [1,2]. Esta malabsorción de grasa, usualmente iniciada en la infancia, es clave, ya que puede conducir a déficit en el desarrollo fisiológico, debido a la incapacidad de tolerar la grasa de la leche materna o la formulada, apareciendo esteatorrea. Niveles anormales de lipoproteínas o esteatosis hepática, representan formas atípicas de la patología. Las manifestaciones devastadoras incluyen retinopatía, osteopatía, neuropatía, y coagulopatía, debido a deficiencias de vitaminas A, D, E y K, respectivamente, las cuales requieren lipoproteínas para su transporte. Teniendo en cuenta que no existe tratamiento para la ABL, aparte de suministrar grandes dosis de vitaminas liposolubles, es fundamental realizar un diagnóstico precoz y manejo dietético optimo, que pueda mejorar notablemente la condición general del paciente [3].
Materiales y métodos
La estrategia de búsqueda y los métodos de selección de estudios se realizó con base en elementos de la declaración PRISMA y guías Cochrane, utilizando términos de búsqueda tales como “Abetalipoproteinemia” y “Bioquímica”, además de sinónimos, los cuales fueron combinados con los operadores “AND” y “OR”, en las bases de datos PubMed, Science Direct, Clinical Key y Ebsco. Los criterios de inclusión, fueron estudios consistentes en revisiones sistemáticas de la literatura, reportes de caso e investigaciones originales, enfocados en la descripción clínico-molecular de la entidad en cualquier grupo etario. Como criterios de no inclusión, solamente se estableció que aquellos estudios con año de publicación por debajo del límite inferior (2000), y aquellos con contenido de conceptos de preparación y acción sin protocolo establecido, no serían tomados en cuenta. Considerando la particularidad del tema, y la poca variedad de publicaciones, se incluyeron estudios escritos tanto en español como en inglés entre los años 2003 y 2019, citando algunas publicaciones más antiguas con valor científico excepcional. La investigación se realizó durante un periodo de 3 meses. Se identificaron 560 artículos potencialmente relevantes, se revisó título y resumen de todos, y fueron seleccionados 480 de los cuales finalmente se incluyeron 54 artículos, posterior a la discriminación en función de los criterios de inclusión y no inclusión.
Etiología
La ABL se presenta debido a una mutación en el gen MTTP, con patrón homocigoto autosómico recesivo. Hasta el momento, hay descritas más de ٣٣ mutaciones que alteran la formación de la proteína microsomal transferidora de triglicéridos (MTP), cuya función es la lipidación inicial de apoB-48 en el intestino y apoB-100 en el hígado, que conduce a la producción de pre-quilomicrones y lipoproteínas de muy baja densidad, respectivamente [2]. El cuadro sintomático de la enfermedad es el resultado de una deficiencia severa de ácidos grasos esenciales y vitaminas liposolubles, especialmente de vitamina E. Suele presentarse al inicio en los infantes como esteatorrea, y distensión abdominal [4]. Si se evalúa el perfil lipídico plasmático en esta condición, se evidenciará hipocolesterolemia extremadamente grave e hipotrigliceridemia debido a la marcada deficiencia de lipoproteínas que contienen apoB [5,6].
No se debe confundir con las hipobetalipoproteinemias (HBL) monogénicas primarias, de las cuales se conocen mutaciones de genes específicos (APOB, PCSK9, SAR1B y ANGPTL3), aunque se especula de otros no identificados [5,7,8]. Las HBL monogénicas pueden tener un patrón de herencia dominante o recesivo. El fenotipo bioquímico de estas se particulariza por la presencia de niveles plasmáticos disminuidos de colesterol total (TC), LDL-C y apoB, por debajo del percentil 5 de distribución en la población [9,10].
Otra entidad muy similar, es la enfermedad de retención de quilomicrones (ERQ), también llamada enfermedad de Anderson, causada por mutaciones bialélicas en el gen SAR1B, que codifica para la Sar1b GTPasa, una trifosfatasa de guanosina, que en los enterocitos interviene en el desdoble de la proteína II (COPII), la cual asiste en el transporte vesicular desde el retículo endoplasmático hasta el aparato Golgi, recubriendo a los pre-quilomicrones. Aquí, la formación de pre-quilomicrones es completamente normal, sin embargo, su maduración y transporte a través de los enterocitos, están alteradas. La hipocolesterolemia en esta condición suele ser más leve que en la ABL y se asocia a niveles normales de triglicéridos plasmáticos [5,8,11].
Otra condición en particular destacable dentro de este complejo de trastornos de mala absorción, similares a la ABL, es la hipobetalipoproteinemia familiar tipo 1, en la cual el gen afectado es el APOB, produciendo malabsorción de lípidos, generalmente en portadores de mutaciones bialélicas de este gen, cohibiendo específicamente la formación de apoB-48, principal componente proteico de los quilomicrones [5,8,9].
Epidemiología
No se tienen registros fiables de esta condición, aunque se describe que es extremadamente rara, presentándose en menos de 1 de cada millón de personas; incluso se describen menos de 100 casos en la literatura mundial [12]. En Colombia no existen datos, lo que supone poco conocimiento sobre esta condición, dificultando así un diagnóstico eficaz, e incluso, tomarlo como diagnóstico diferencial. Los productos de matrimonios consanguíneos, son un factor de riesgo inminente. Teniendo en cuenta su patrón de heredabilidad, ambas copias del gen deben estar alteradas para la presentación de la patología. Su distribución en hombres y mujeres es igual. Existe una mutación en particular en las personas de descendencia judía ashkenazi, localizada en el centro de Europa. Dicha mutación reemplaza el aminoácido glicina durante el proceso de traducción, generando una señal de parada en la posición 865 (Gly865X o G865X). Resultado de estos cambios, se elabora una versión anormalmente pequeña y disfuncional de la proteína [12,13]
Fisiopatología
Las beta-apolipoproteínas son una subfamilia de apolipoproteínas de gran tamaño; son esenciales para la secreción y formación de quilomicrones y VLDL. MTP actúa como un auxiliar que facilita la transferencia de lípidos a apoB [14]. Esta se encuentra dentro de los microsomas en el tejido hepático y la mucosa intestinal y cumple como función principal la catálisis de transferencia de triglicéridos, ésteres de colesterol, y fosfatidilcolina entre membranas. El flujo de transporte de lípidos disminuye en el orden de triglicéridos - ésteres de colesterol - diglicérido - colesterol - fosfatidilcolina [15,16]. MTP es un heterodimero que contiene subunidades de 58 y 97 kDa, siendo esta última, la que posee la actividad de transferencia de lípidos. Esta subunidad, puede faltar en la ABL [1,17].
El proceso comienza en el retículo endoplasmático, generándose las apolipoproteínas, colesterol, fosfolípidos y triglicéridos e incorporándose luego en partículas de lipoproteínas. Estos complejos se trasladan al aparato de Golgi y se secretan [18]. Estas lipoproteínas son específicas en su composición lipídica y tipo de apolipoproteínas que contiene. Las dos beta-apolipoproteínas que se originan, son B-48 y B-100, incluyéndose esta última en las VLDL [19]. ApoB-100 se sintetiza en el tejido hepático, y a diferencia de apoB-48, posee el sitio de unión particular para la unión de LDL con receptores LDL en hepatocitos [20,21].
El mecanismo fisiopatológico se basa en la ausencia del heterodimero de disulfuro de la proteína MTP, que actúa como chaperona para el origen del complejo apoB-lipoproteína [22]. Este es primordial para el proceso de lipidación del complejo naciente [23]. En caso de deficiencia de MTP, apoB no puede incluir lípidos neutros, y es degradado por los proteosomas. Producto de todo este proceso, se da la alteración en la secreción de quilomicrones y VLDL del intestino y el hígado respectivamente [24,25]. Esta carencia de compuestos, conducen a la malabsorción de grasas y subsiguiente desarrollo de la sintomatología [26,27]. La privación en la secreción de VLDL, ocasiona esteatosis hepática que puede avanzar a cirrosis, por el proceso fibrótico [20,28,29].
Manifestaciones gastrointestinales
Son las primeras en manifestarse, y las que generan la alerta del cuadro; se presentan en la infancia, y consisten principalmente en diarrea o esteatorrea y deficiencia de vitaminas liposolubles. Es extraño, pero la diarrea puede no ser un síntoma evidente, puesto que al observar la exacerbación del cuadro con alimentos grasos, muchas veces se evitan, no acudiendo a asistencia médica y ocultando el trastorno, lo que puede dificultar el diagnóstico precoz. A pesar de lo anterior, el déficit de vitaminas liposolubles continúa intacto, ya que su asimilación y transporte dependen esencialmente de la vía metabólica [30,31]. La severidad de la sintomatología se relaciona directamente con la cantidad de grasa consumida; por lo tanto, si se aprende a evitar la grasa dietética, mejora la esteatorrea [32,33].
Manifestaciones hepáticas
A veces los marcadores hepáticos pueden durar estables por muchos años, dándose el caso en el que ni siquiera evolucione significativamente en la clínica. Pero en caso tal, se puede evidenciar elevación de las transaminasas, hepatomegalia y esteatosis hepática, existiendo la posibilidad de trascender a esteatohepatitis, fibrosis y muy inusualmente a cirrosis [33]. Si se mantiene una dieta típica sin restricción lipídica, durante una endoscopia, se podrá observar el signo de “escarcha de ron blanco” en la mucosa intestinal. No obstante, existen reportes de paciente con cirrosis [34,35].
Manifestaciones hematológicas
La acantocitosis es esencial en la distinción del cuadro patológico. Los acantocitos son eritrocitos con una estructura irregular, generalmente puntiforme, producto de una membrana celular defectuosa por carencia de fosfolípidos. También se ven en la disfunción hepática. Esta condición de malabsorción podría conducir a deficiencias de hierro o ácido fólico, finalizando así en anemia. También puede presentarse hemolisis debido a incremento en la peroxidación lipídica por deficiencia de la vitamina E, ya que esta participa en vías de protección contra el estrés oxidativo y se observaría exacerbación de la anemia [33,35]. Otro dato importante es el rol de la vitamina K en el proceso fisiopatológico, puesto que participa en la descarboxilación de los factores de coagulación; cuando hay carencia se presentan trastornos hemorrágicos, disminución de la tasa de sedimentación de eritrocitos, exacerbación aún más de la anemia, reticulocitosis, hiperbilirrubinemia, y un INR prolongado [31,36].
Manifestaciones neurológicas
Esta sintomatología suele ser confusa, por el gran espectro de diagnósticos diferenciales; sin embargo, deben ser analizados de forma exhaustiva, debido al grado de variabilidad y severidad de la afectación del sistema nervioso central y periférico. Principalmente se presenta neuropatía periférica, trastorno del desarrollo intelectual, temblor, nistagmo, disminución de los reflejos tendinosos profundos o arreflexia. La fisiopatología se basa en la desmielinización, a causa de la deficiencia de vitaminas liposolubles [31,37,38].
Manifestaciones ópticas
Estas son variables, sin embargo, la más característica es la pigmentación atípica de la retina. Un gran volumen de individuos afectados permanecen asintomáticos hasta avanzada edad, cuando desarrollan pérdida de la visión y/o visión de color. Durante la evolución de esta enfermedad se puede experimentar escotomas en expansión progresiva [39]. Si no se detecta y trata de forma precoz, puede producirse ceguera rápidamente. Otros hallazgos raros pueden ser ptosis, oftalmoplejia o úlceras corneales. Se ha descrito que la ptosis y la oftalmoplejía se producen debido a la desmielinización del nervio craneal por deficiencia de vitamina E. Las úlceras corneales se producen o exacerban por la deficiencia de vitamina A [30]. También pueden ser signos de alarma la retinitis pigmentosa o disminución de la visión nocturna [31,40,41,42].
Manifestaciones musculares
Las manifestaciones neuromusculares suelen aparecer en la primera o segunda década de vida, secundario a la deficiencia de vitamina E. Los síntomas más frecuentes son la pérdida progresiva de reflejos tendinosos profundos, sentido vibratorio y propiocepción, hipotonía, disartria, y muy frecuentemente, una ataxia muy parecida a la de Friedrich, con una marcha escalonada alta, que puede comenzar a principios de edad adulta en individuos sin manejo hasta ese momento [31,43,44].
Manifestaciones cardiacas y endocrinológicas
Son de muy rara presentación, sin embargo, se han reportado casos de cardiomegalia en la edad adulta, con muerte súbita, sin etiología específica. También se han notificado casos de hipotiroidismo subclínico [45,46,47].
Diagnóstico
Básicamente el diagnóstico es clínico, bioquímico y molecular; primero se evalúan los niveles de LDL, colesterol, triglicéridos, apo-B y vitaminas liposolubles, los cuales deben estar ausentes o extremadamente bajos [31]. Si la evaluación del fenotipo y laboratorio, son compatibles con la entidad, se realizan pruebas genéticas moleculares en busca de alteraciones un solo gen o un panel multigén. El análisis de la secuencia de MTTP puede detectar pequeñas deleciones o inserciones intragénicas, y variantes de sitio de cambio de sentido o empalme. Es excepcional encontrar deleciones/duplicaciones de un exón o de genes enteros [1].
Existe literatura que recomienda utilizar solamente el panel multigénico, ya que tiene más probabilidad de identificar la alteración genética, puesto que el panel incluye a MTTP y otros genes de interés simultáneamente; además, sería más costo-efectivo y limitaría la identificación de variantes de importancia incierta y variantes patógenas en genes que no explican el fenotipo presentado [31].
Con el fin de encontrar probables signos ocultos se debe hacer un recuento de sangre completo, para identificar anemia, trombocitopenia o pancitopenia. También se debe hacer un frotis sanguíneo, que nos mostraría acantocitosis (células de Burr); o estudiar las heces, que revelarían la malabsorción de grasas y estudios de imágenes, para explorar integridad hepática o ultrasonografía para evaluar cambios en la esteatosis. La resonancia magnética podría ayudar a identificar degeneración de la región espinocerebelosa, en caso de existir. El examen ocular y de retina comprobaría si existe lesión en este órgano [48,49,50,51,52].
En general, si el cuadro es típico, con manifestaciones clásicas de retraso en el desarrollo, diarrea, vómitos, esteatorrea, hepatomegalia, pérdida o disminución de visión nocturna y/o de color, pigmentación atípica adquirida de la retina o ataxia espinocerebelosa, se debe sospechar inmediatamente en ABL [53,54].
Diagnósticos diferenciales
Existe un amplio número de diagnósticos diferenciales (Tabla 1) los cuales deben ser descartados por un equipo multidisciplinario. Ya que esta condición se describe como un síndrome de malabsorción, hay que tomar en cuenta otros tales como hipobetalipoproteinemias monogénicas primarias, hipobetalipoproteinemia familiar tipo 1, y la enfermedad de retención de quilomicrones. Del mismo modo, se pueden considerar otras como la enfermedad hepática colestásica crónica, neuropatía combinada y atáxica, deficiencia familiar de vitamina E, neuropatías sensitivomotoras hereditarias, degeneración de la retina y trastornos espinocerebelosos [31]. Por tanto, se debe ser muy exhaustivos al establecer el diagnóstico, debido a que la clínica no es propia de la entidad, dando una sintomatología inespecífica, más aún, si se presenta un cuadro atípico. Sin embargo, los estudios bioquímicos y moleculares, juegan un rol fundamental en este proceso.
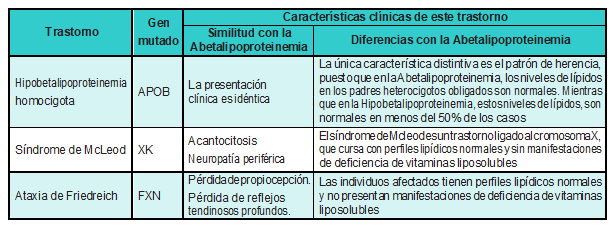 Tabla 1
Principales trastornos a considerar en el diagnóstico diferencial de la Abetalipoproteinemia [31]
Tabla 1
Principales trastornos a considerar en el diagnóstico diferencial de la Abetalipoproteinemia [31]
Abordaje terapéutico
Las principales recomendaciones pretenden disminuir la sintomatología y prevenir complicaciones;por lo menos al instaurar el tratamiento precoz con vitamina E se puede retrasar e incluso prevenir trastornos en el desarrollo neurológico, así como oftalmoplejia o ptosis palpebral. Igual sucede cuando se implanta el tratamiento con vitamina A que puede prevenir el desarrollo del úlceras corneales [31,34] (Tabla 2).
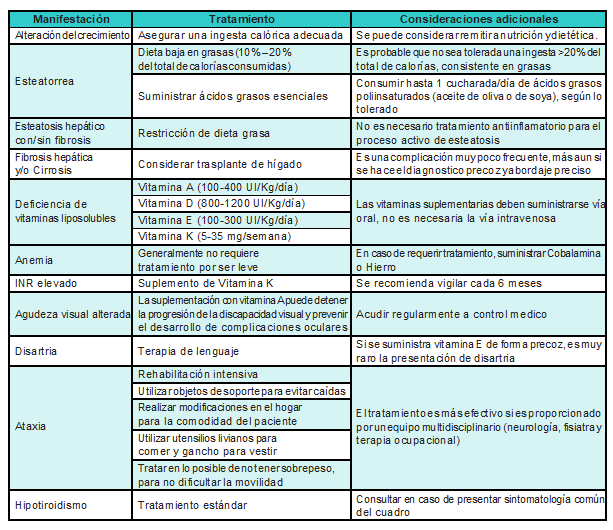 Tabla 2
Tratamiento sintomático de la Abetalipoproteinemia [31]
Tabla 2
Tratamiento sintomático de la Abetalipoproteinemia [31]
INR: International Normalized Ratio
También se debería brindar el servicio de asesoramiento genético, el cual permite asumir de forma más plena la situación, al otorgar una información completa sobre el proceso salud-enfermedad, y definir un plan de reproducción puntual, que confiera la posibilidad de realizar test prenatal para diagnósticos durante la gestación. Además, al brindar asesoría sobre la situación, se adquiere el conocimiento de poseer un alelo afectado, que será tomado en cuenta a la de plantear la opción de generar descendencia, así como el riesgo de afectación [31].
En un tratamiento adecuado puede lograr un patrón de crecimiento normal en función del tiempo, pero no garantiza un crecimiento total hasta las edades más avanzadas del desarrollo. Otra recomendación es que al individualizar la dieta baja en lípidos, debe evitarse totalmente el consumo de ácidos grasos de cadenas largas. También es importante recalcar que la dosis de vitamina A se debe ajustar teniendo en cuenta la concentración seria de beta caroteno, planteándose como objetivo mantener estas concentraciones sobre el límite inferior, para evitar hepatotoxicidad. A pesar de la suplementación, estos individuos siempre permanecerán con niveles de vitamina E por debajo de los niveles normales [31].
Perspectivas futuras
Liu et al. realizaron un estudio con células madre pluripotenciales inducidas, generadas a partir de un paciente con ABL para evaluar la expresión y funcionalidad de estas siendo diferenciadas como hepatocitos y cardiomiocitos. Demostraron que los hepatocitos y cardiomiocitos humanos exhiben defectos asociados con la ABL, debido a la pérdida de apolipoproteína B, además de la secreción y acumulación intracelular de lípidos. Sin embargo, el dato a enfatizar fue que posterior a la corrección de la mutación por edición genética CRISPR / Cas9, este fenotipo fue revertido [28]. Esto permite afirmar que existe la posibilidad de estipular un tratamiento complejo para corregir esta alteración y proporcionar una mejor calidad de vida para los individuos afectados por esta patología.
Conclusión
A pesar de que la ABL es un trastorno de presentación excepcional, nunca estamos exonerados de encontrarnos ante un nuevo caso, por lo que es necesario conocer sus características básicas, sus manifestaciones clásicas o atípicas, la importancia de hacer un diagnóstico precoz y abordaje preciso y multidisciplinar. De la misma forma, resaltar que hace parte de un grupo de síndromes de malabsorción, y de ahí la importancia de realizar los diagnósticos diferenciales. Tener claro que en caso de no establecer un manejo adecuado, las complicaciones son muchas y severas. Desafortunadamente en Colombia no existe registro alguno sobre esta condición, por lo que el manejo en base a guías de práctica clínica de sociedades nacionales es nulo, esto demuestra que es necesario intensificar el diseño de políticas en materia de salud pública, para la investigación de enfermedades raras a nivel nacional.
Conflicto de interés: los autores declaran no tener conflicto de interés.
Fuentes de financiación: el artículo fue financiado por los autores.
Literatura citada
1. Boltshauser E, Weber KP. Laboratory investigations. Handb Clin Neurol 2018; 154:287-298. DOI: 10.1016/B978-0-444-63956-1.00017-5
2. Mushtaq I, Cheema HA, Malik HS, Waheed N, Hashmi MA, Malik HS. Causes Of Chronic Non-Infectious Diarrhoea In Infants Less Than 6 Months Of Age: Rarely Recognized Entities. J Ayub Med Coll Abbottabad 2017; 29(1):78-82.
3. Ueda M, Burke F, Maeda M, McIntyre A, Hegele R, Malloy M, et al. Importance of Nutritional Intervention for Infants with Abetalipoproteinemia. J Clin Lipidol 2019; 13(3):e44. DOI: 10.1016/j.jacl.2019.04.074
4. Strain JE, Vigilante JA, DiGeorge NW. Hypolipidemia in a Special Operations Candidate: Case Report and Review of the Literature. J Spec Oper Med 2015; 15(4):1-5.
5. Tarugi P, Averna M. Hypobetalipoproteinemia: genetics, biochemistry, and clinical spectrum.Adv Clin Chem 2011; 54:81–107. DOI: https://doi.org/10.1016/B978-0-12-387025-4.00004-2
6. Simone ML, Rabacchi C, Kuloglu Z, Kansu A, Ensari A, Demir AM, et al. Novel mutations of SAR1B gene in four children with chylomicron retention disease. J Clin Lipidol 2019; 13(4):554-562. DOI: 10.1016/j.jacl.2019.05.013
7. Kersten S. Angiopoietin-like 3 in lipoprotein metabolism. Nat Rev Endocrinol 2017; 13(12):731–739. DOI: 10.1038/nrendo.2017.119
8. Levy E. Insights from human congenital disorders of intestinal lipid metabolism. J Lipid Res 2015; 56(5):945–962. DOI: 10.1194/jlr.R052415
9. Julve J, Martin-Campos JM, Escola-Gil JC, Blanco-Vaca F. Chylomicrons: advances in biology, pathology, laboratory testing, and therapeutics. Clin Chim Acta 2016; 455:134–148. DOI: 10.1016/j.cca.2016.02.004
10. Schonfeld G, Lin X, Yue P. Familial hypobetalipoproteinemia: genetics and metabolism. Cell Mol Life Sci 2005; 62(12):1372–1378. DOI: https://doi.org/10.1007/s00018-005-4473-0
11. Peretti N, Sassolas A, Roy CC, Deslandres C, Charcosset M, Castagnetti J, et al. Guideline for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers.Orphanet J Rare Dis 2010; 5:24. DOI: 10.1186/1750-1172-5-24.
12. Hentati F, El-Euch G, Bouhlal Y, Amouri R. Ataxia with vitamin E deficiency and abetalipoproteinemia. Handb Clin Neurol 2012; 103:295-305. DOI: 10.1016/B978-0-444-51892-7.00018-8.
13. Wang LR, McIntyre AD, Hegele RA. Complex genetic architecture in severe hypobetalipoproteinemia. Lipids Health Dis 2018; 17(1):48. DOI: 10.1186/s12944-018-0680-1
14. Ramasamy I. Update on the molecular biology of dyslipidemias. Clin Chim Acta 2016; 454:143-85. DOI: 10.1016/j.cca.2015.10.033
15. Yilmaz BS, Mungan NO, Di Leo E, Magnolo L, Artuso L, Bernardis I, et al. Homozygous familial hypobetalipoproteinemia: A Turkish case carrying a missense mutation in apolipoprotein B. Clin Chim Acta 2016; 452:185-190. DOI: 10.1016/j.cca.2015.11.017
16. Walsh MT, Iqbal J, Josekutty J, Soh J, Di Leo E, Özaydin E, et al. Novel Abetalipoproteinemia Missense Mutation Highlights the Importance of the N-Terminal β-Barrel in Microsomal Triglyceride Transfer Protein Function. Circ Cardiovasc Genet 2015; 8(5):677-687. DOI: 10.1161/CIRCGENETICS.115.001106
17. Di Filippo M, Frachon S, Janin A, Rajan S, Marmontel O, Decourt C, et al. Normal serum ApoB48 and red cells vitamin E concentrations after supplementation in a novel compound heterozygous case of abetalipoproteinemia.Atherosclerosis 2019; 284:75-82. DOI: 10.1016/j.atherosclerosis.2019.02.016
18. Cuerq C, Henin E, Restier L, Blond E, Drai J, Marçais C, et al. Efficacy of two vitamin E formulations in patients with abetalipoproteinemia and chylomicron retention disease. J Lipid Res 2018; 59:1640–1648. DOI: 10.1194/jlr.M085043
19. Hooper AJ, Burnett J. Update on primary hypobetalipoproteinemia. Curr Atheroscler Reports 2014; 16(7):423. DOI: 10.1007/s11883-014-0423-3
20. Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia.J Hepatol 2014; 61:891–902. DOI: 10.1016/j.jhep.2014.05.023
21. Sirwi A, Hussain MM. Lipid transfer proteins in the assembly of apoB-containing lipoproteins. J Lipid Res 2018; 59:1094-1102. DOI: 10.1194/jlr.R083451
22. Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly.J Lipid Res 2003; 44:22-32. DOI: 10.1194/jlr.R200014-JLR200
23. Davis RA, Thrift RN, Wu CC, Howell KE. Apolipoprotein B is both integrated into and translocated across the endoplasmic reticulum membrane. Evidence for two functionally distinct pools. J Biol Chem 1990; 265(17):10005-10011.
24. Zhou M, Fisher EA, Ginsberg HN. Regulated Co-translational ubiquitination of apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein.J Biol Chem 1998; 273:24649-24653.
25. Miller SA, Burnett JR, Leonis MA, McKnight CJ, van Bockxmeer FM, Hooper AJ. Novel missense MTTP gene mutations causing abetalipoproteinemia. Biochim Biophys Acta 2014; 1841(10):1548-1554. DOI: 10.1016/j.bbalip.2014.08.001
26. Gündüz M, Özaydın E, Atar MB, Koç N, Kırsaçlıoğlu C, Köse G, et al., Microsomal triglyceride transfer protein gene mutations in Turkish children: A novel mutation and clinical follow up. Indian J Gastroenterol 2016; 35(3):236-241. DOI: 10.1007/s12664-016-0654-z
27. Paquette M, Dufour R, Hegele RA, Baass A. A tale of 2 cousins: An atypical and a typical case of abetalipoproteinemia.J Clin Lipidol 2016; 10(4):1030-1034. DOI: 10.1016/j.jacl.2016.01.003
28. Liu Y, Conlon DM, Bi X, Slovik KJ, Shi J, Edelstein HI, et al., Lack of MTTP Activity in Pluripotent Stem Cell-Derived Hepatocytes and Cardiomyocytes Abolishes apoB Secretion and Increases Cell Stress. Cell Rep 2017; 19(7):1456-1466. DOI: 10.1016/j.celrep.2017.04.064
29. Di Filippo M, Varret M, Boehm V, Rabés JP, Ferkdadji L, Abramowitz L, et al. Post-prandial lipid absorption in seven heterozygous carriers of deleterious variants of MTTP in two abetalipoproteinemic families. J Clin Lipidol 2018; 13(1):201-212. DOI: 10.1016/j.jacl.2018.10.003
30. Shoulders CC, Brett DJ, Bayliss JD, Narcisi TM, Jarmuz A, Grantham TT, et al. Abetalipoproteinemia is caused by defects of the gene encoding the 97 kDa subunit of a microsomal triglyceride transfer protein. Hum Mol Genet 1993; 2:2109-2116. DOI: https://doi.org/10.1093/hmg/2.12.2109
31. Burnett JR, Hooper AJ, Hegele RA. Abetalipoproteinemia En: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviws. Seattle: University of Washington; 2018.
32. Khatun I, Walsh MT, Hussain MM. Loss of both phospholipid and triglyceride transfer activities of microsomal triglyceride transfer protein in abetalipoproteinemia. J Lipid Res 2013; 54:1541–1549. DOI: 10.1194/jlr.M031658
33. Lee J, Hegele RA. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J Inherit Metab Dis 2014; 37:333–339. DOI: 10.1007/s10545-013-9665-4
34. Zamel R, Khan R, Pollex RL, Hegele RA. Abetalipoproteinemia: two cases and literature review. Orphanet J Rare Dis 2008; 3:19. DOI: 10.1186/1750-1172-3-19
35. Aers XP, Leroy BP, Defesche JC, Shadid S. Abetalipoproteinemia From Previously Unreported Gene Mutations. Ann Intern Med 2019; 170(3):211-213. DOI: 10.7326/L18-0358
36. Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond) 2012; 9:14. DOI: 10.1186/1743-7075-9-14
37. Hammer MB, El Euch-Fayache G, Nehdi H, Feki M, Maamouri-Hicheri W, Hentati F, et al. Clinical features and molecular genetics of two Tunisian families with abetalipoproteinemia. J Clin Neurosci 2014; 21(2):311–315. DOI: 10.1016/j.jocn.2013.04.016
38. Chardon L, Sassolas A, Dingeon B, Michel-Calemard L, Bovier-Lapierre M, Moulin P, et al. Identification of two novel mutations and long-term follow-up in abetalipoproteinemia: a report of four cases. Eur J Pediatr 2009; 168(8):983–989. DOI: 10.1007/s00431-008-0888-6
39. Najah M, Youssef SM, Yahia HM, Afef S, Awatef J, Saber H, et al. Molecular characterization of Tunisian families with abetalipoproteinemia and identification of a novel mutation in MTTP gene. Diagn Pathol 2013; 48:54. DOI: 10.1186/1746-1596-8-54
40. Zeissig S, Dougan SK, Barral DC. Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function.J Clin Invest 2010; 120:2889–99. DOI: 10.1172/JCI42703
41. Barakizou H, Gannouni S, Messaoui K, Difilippo M, Sassolas A, Bayoudh F. Abetalipoproteinemia: A novel mutation of microsomal triglyceride transfer protein (MTP) gene in a young Tunisian patient.Egypt J Med Hum Genet 2016; 17(3):251-254. DOI: 10.1016/j.ejmhg.2015.12.003
42. Rashtian P, Najafi Sani M, Jalilian R. A Male Infant with Abetalipoproteinemia: A Case Report from Iran. Middle East J Dig Dis 2015; 7:181-4.
43. Pons V, Rolland C, Nauze M, Danjoux M, Gaibelet G, Durandy A, et al. A severe form of Abetalipoproteinemia caused by new splicing mutations of microsomal triglyceride transfer protein (MTTP). Human Mutation 2011; 32:751–759. DOI: 10.1002/humu.21494
44. Uslu N, Gurakan F, Yuce A, Demir H, Tarugi P. Abetalipoproteinemia in an infant with severe clinical phenotype and a novel mutation. Turk J Pediat 2010; 52(1):73-77.
45. Burnett JR, Hooper AJ. Vitamin E and oxidative stress in abetalipoproteinemia and familial hypobetalipoproteinemia. Free Radic Biol Med 2015; 88:59-62. DOI: 10.1016/j.freeradbiomed.2015.05.044
46. Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol 2014; 25(3):161–168. DOI: 10.1097/MOL.0000000000000072
47. Burnett JR, Zhong S, Jiang ZG, Hooper AJ, Fisher EA, McLeod RS, et al. Missense mutations in APOB within the betaalpha1 domain of human APOB-100 result in impaired secretion of apoB and apoB-containing lipoproteins in familial hypobetalipoproteinemia.J Biol Chem 2007; 282(33):24270–24283. DOI: https://doi.org/10.1074/jbc.M702442200
48. Tanoli T, Yue P, Yablonskiy D, Schonfeld G. Fatty liver in familial hypobetalipoproteinemia: roles of the APOB defects, intra-abdominal adipose tissue, and insulin sensitivity.J Lipid Res 2004; 45(5):941–947. DOI: 10.1194/jlr.M300508-JLR200
49. Traber MG. Mechanisms for the prevention of vitamin E excess. J Lipid Res 2013; 54(9):2295–2306. DOI: 10.1194/jlr.R032946
50. Ulatowski L, Manor D. Vitamin E trafficking in neurologic health and disease.Annu Rev Nutr 2013; 33:87–103. DOI: 10.1146/annurev-nutr-071812-161252
51. Havel RJ, Kane JP. Introduction: structure and metabolism of plasma lipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 7th ed. New York: McGraw-Hill; 1995. p. 1841-1851. DOI: 10.1036/ommbid.142
52. Young SG. Recent progress in understanding apolipoprotein B. Circulation 1990; 82(5):1574–1594. DOI: https://doi.org/10.1161/01.CIR.82.5.1574
53. Linton MF, Farese RV Jr, Young SG. Familial hypobetalipoproteinemia. J Lipid Res 1993; 34(4):521–541.
54. Visser ME, Lammers NM, Nederveen AJ, Van der Graaf M, Heerschap A, Ackermans MT, et al. Hepatic steatosis does not cause insulin resistance in people with familial hypobetalipoproteinaemia. Diabetologia 2011; 54:2113–2121. DOI: 10.1007/s00125-011-2157-x
