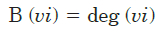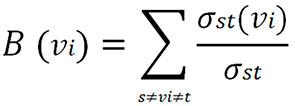RESUMEN
Introducción: La restricción del crecimiento intrauterino es una alteración del desarrollo fetal que se caracteriza por una tasa de crecimiento durante la etapa fetal que es menor al potencial genético de crecimiento para la edad gestacional. Esta condición plantea una carga importante para la salud pública, ya que aumenta la morbimortalidad de la descendencia, a corto y a largo plazo, particularmente, por asociarse al desarrollo de enfermedad cardiovascular y metabólica en la vida adulta.
Objetivos: Mediante el uso de herramientas bioinformáticas nos propusimos identificar posibles genes cardinales involucrados en la restricción del crecimiento intrauterino asociados al desarrollo de obesidad, hipertensión arterial y síndrome metabólico.
Material y métodos: Obtuvimos un total de 343 genes involucrados en los fenotipos de interés e identificamos 20 genes que resultaron significativamente relevantes en el análisis de la red de interacción. Particularmente, cuatro de estos genes identificados codifican para factores de crecimiento o sus receptores, VEGFA, PDGFRB, IGF1R y EGFR. Además, identificamos genes relacionados con la insulina y el control de la homeostasis cardiovascular, como son el CTNNB1, APP, MYC y MDMD2. Por otra parte, el análisis de clústeres permitió reconocer los términos de ontología genética más significativos, entre los que se destacan aquellos relacionados con procesos biológicos de proliferación y muerte celular programada, de comunicación intercelular, del metabolismo proteico, y de desarrollo del sistema cardiovascular.
Conclusiones: Los genes hallados en este estudio podrían ser de utilidad como biomarcadores putativos de la presencia de alteraciones cardiovasculares y metabólicas asociadas a la restricción del crecimiento intrauterino o potenciales blancos terapéuticos de estrategias de tratamiento orientadas al genotipo del paciente.
Palabras claves: Biología computacional, Ontología de genes, Retardo del crecimiento fetal, Hipertensión, Obesidad, Síndrome metabólico.
ABSTRACT
Background: Intrauterine growth restriction is an abnormal fetal development characterized by a fetal growth rate lower than the potential genetic growth for the gestational age. This condition represents a major burden for public health systems, as it increases short and long-term morbidity and mortality in the offspring, particularly because of its association with the development of cardiovascular and metabolic disease in adult life.
Objectives: The aim of the present study was to identify possible cardinal genes involved in intrauterine growth restriction associated with the development of obesity, hypertension and metabolic syndrome using bioinformatics tools.
Methods: A total of 343 genes involved in the phenotypes of interest were obtained and 20 genes were identified as significantly relevant in the interaction network analysis. Specifically, four of these identified genes encode for growth factors or their receptors, VEGFA, PDGFRB, IGF1R and EGFR. We also identified genes related to insulin and cardiovascular homeostasis as CTNNB1, APP, MYC and MDMD2. Cluster analysis provided the most significant gene ontology terms, including those related to the biological processes of proliferation and programmed cell death, intercellular communication, protein metabolism and development of the cardiovascular system.
Conclusions: The genes found in this study could be useful as putative biomarkers for the presence of cardiovascular and metabolic disorders associated with intrauterine growth restriction, or as potential therapeutic targets for treatment strategies directed to the patient’s genotype.
Key words: Computational Biology, Gene Ontology, Fetal Growth Retardation, Hypertensión, Obesity, Metabolic Syndrome.
Carátula del artículo
Programación intrauterina de enfermedad en la vida adulta: identificación de genes cardinales asociados a hipertensión arterial, obesidad y síndrome metabólico
Intrauterine Programming of Adult Disease: Identification of Cardinal Genes Associated with Hypertension, Obesity and Metabolic Syndrome
 IVÁN D ACEVEDO MONTERROSA
IVÁN D ACEVEDO MONTERROSA
Universidad de Buenos Aires, Argentina
DAMIÁN A. SORIA
Universidad de Buenos Aires, Argentina
ANALÍA TOMAT
Universidad de Buenos Aires, Argentina
Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina
ROSANA ELESGARAY
Universidad de Buenos Aires, Argentina
Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina
CRISTINA ARRANZ
Universidad de Buenos Aires, Argentina
Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina
CAROLINA CANIFFI
Universidad de Buenos Aires, Argentina
Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina
Revista argentina de cardiología, vol. 89, núm. 1, pp. 27-36, 2021
Sociedad Argentina de Cardiología
Recepción: 24 Noviembre 2020
Aprobación: 08 Enero 2021
Financiamiento
Fuente: Universidad de Buenos Aires
Nº de contrato: UBACYT 20020170200093BA, 2018-2019; 20020190200227BA, 2020-2021
Descripción del financiamiento: Este trabajo fue financiado por la Universidad de Buenos Aires (UBA) [UBACYT 20020170200093BA, 2018-2019; 20020190200227BA, 2020-2021]; y el Instituto de Química y Metabolismo del Fármaco (IQUIMEFA), UBA-CONICET, Argentina
INTRODUCCIÓN
El desarrollo durante el período prenatal es un proceso altamente dinámico en el que se combinan factores maternos, placentarios y fetales, junto con factores genéticos y ambientales, que pueden modular la proliferación y maduración celular, durante las etapas embrionaria y fetal del crecimiento intrauterino. 1,2
La restricción o retardo del crecimiento intrauterino (RCIU) es una alteración del desarrollo fetal que puede estar presente en aproximadamente el 5% al 15% de los niños nacidos vivos, 3,4 y que se caracteriza por una tasa de crecimiento durante la etapa fetal que es menor que el potencial genético de crecimiento para la edad gestacional. 2 Esta complicación durante el embarazo plantea una carga importante para los sistemas de salud, ya que aumenta la morbilidad y la mortalidad de la descendencia, tanto a corto como a largo plazo. 5,6
En la década de 1980, Barker postuló que la enfermedad cardiovascular en el adulto podría tener sus orígenes en la vida fetal con base en la observación de la distribución geográfica de la mortalidad infantil a principios del siglo XX, que era similar a la de muerte por cardiopatía isquémica aproximadamente 60 años después. 7 Desde entonces, se han realizado numerosos estudios epidemiológicos, clínicos y experimentales, que permitieron profundizar el conocimiento de los factores que pueden afectar negativamente el desarrollo durante el período fetal, entre ellos la malnutrición materna, la hipertensión gestacional, la preeclampsia/ eclampsia, la diabetes gestacional, la menor actividad del sistema renina angiotensina en los riñones, y la exposición a hipoxia o estrés. 2 En particular, numerosos estudios han apoyado la hipótesis de la programación temprana de la enfermedad cardiovascular en la vida adulta; el aporte de nutrientes al feto es una de las principales causas. 8,9,10,11 Además, en un esfuerzo por conservar energía y sobrevivir a expensas de completar el crecimiento fetal en un entorno de suministro inadecuado de nutrientes, se ha observado la alteración de la homeostasis de la glucosa y la insulina fetal, que modifica el perfil metabólico en los tejidos, y programa la expresión de adaptaciones en la vida posnatal asociadas a la resistencia a la insulina y al exceso de peso. 12 Más aún, se ha observado que el daño al que se expone a un individuo durante su desarrollo intrauterino puede tener consecuencias metabólicas en las generaciones posteriores. 13,14
En la sociedad moderna, y particularmente en Argentina, existe una alta prevalencia de los factores de riesgo para el desarrollo de las enfermedades cardiovasculares y metabólicas. 15 Por lo tanto, el desarrollo de estrategias de detección temprana de la susceptibilidad de un individuo, que nació con RCIU, a presentar enfermedad cardiovascular o metabólica en la vida adulta podría mitigar el riesgo de consecuencias a largo plazo de la RCIU mediante intervenciones terapéuticas y estrategias de tratamiento orientadas.
Actualmente, contamos con gran cantidad de datos en la bibliografía sobre la RCIU, la hipertensión arterial (HTA), la obesidad y el síndrome metabólico (SM). Este último representa la asociación de varios factores de riesgo cardiovascular entre los que se incluye la obesidad, la HTA, la dislipemia, y la resistencia a insulina. 16 Sin embargo, estos datos de la bibliografía se encuentran fragmentados en diferentes niveles de información biológica (genómica, transcriptómica, proteómica). El procesamiento e integración de datos usando herramientas bioinformáticas nos acerca a la posibilidad de aplicar una medicina personalizada.
Por lo tanto, en el presente trabajo nos propusimos evaluar e identificar posibles genes cardinales involucrados en la RCIU asociados al desarrollo de HTA, obesidad y SM, y los procesos biológicos que puedan estar alterados.
MATERIAL Y MÉTODOS
Recolección de datos
Los genes relacionados con las condiciones de RCIU, obesidad, HTA, y SM, fueron recolectados en julio 2019 a partir de las siguientes bases de datos: Phenotype-Genotype Integrator (PheGenI https://www.ncbi.nlm.nih.gov/gap/phegeni), Gene NCBI (https://www.ncbi.nlm.nih.gov/gene), Clinvar (http:// www.ncbi.nlm.nih.gov/clinvar/), 17 Database of gene-disease associations (DisGeNET https://www.disgenet.org/), 18 European Bioinformatic Institute (EBI http://www.ebi.ac.uk) 19 y Comparative Toxicogenomics Database (CTD http:// ctdbase.org). 20 De esta última, se seleccionaron únicamente los genes con una relación directa con los fenotipos basada en la bibliografía publicada.
Construcción de las redes de interacción genética
Las redes de interacción genética son una representación matemática de la interacción entre genes codificantes de proteínas en sistemas biológicos. Esta relación se establece a través de diferentes métodos, como minería de textos, información basada en experimentos, interacciones establecidas por modelos de ortología, métodos estadísticos, y predicciones basadas en características proteicas. Para la creación de las redes de interacción se emplearon tres herramientas ampliamente usadas: Search Tool for the Retrieval of Interaction Genes/Protein (STRING https://string-db.org/) 21 creada por Swiss Institute of Bioinformatics (SIB) y European Molecular Biology Laboratory (EMBL); Consensus Path DB-Human (CPDB http://cpdb.molgen.mpg.de/), 22 desarrollada por MAX Planck Institute for Molecular Genetics; y Gene MANIA (https://genemania.org/) 23 desarrollada por Donnelly Centre for Cellular and Biomolecular Research en la Universidad de Toronto, Canadá.
Análisis de redes de interacción genética y evaluación topológica
Las redes fueron visualizadas y analizadas en Cytoscape V3. 24 Los genes más relevantes fueron determinados en relación con el grado de centralidad (Degree of centrality) una medida de centralidad topológica que define el número de aristas que inciden en un nodo. Es decir, se utiliza para identificar genes relevantes involucrados directamente con otros genes codificantes de proteínas de la red. La ecuación para calcularlo es la siguiente:
Los genes más relevantes fueron determinados en relación con el grado de centralidad (Degree of centrality), una medida de centralidad topológica que define el número de aristas que inciden en un nodo 25.
Para un gráfico B y un nodo vi, deg representa el número de artistas
Otra propiedad tenida en cuenta fue la intermediación de nodos (Betweeness centrality), que representa cuantitativamente el número de veces que el nodo funciona como un intermediario entre otros nodos. Por lo tanto, esta medida muestra conectores claves para la dinámica de una red biológica. Es calculada usando la siguiente formula: 26
Para un gráfico B, un nodo vi , y dos nodos diferentes a vi (s y t). Donde δst (vi ) es el número de caminos más cortos desde s a t que involucran a vi y δst el camino más corto de s a t.
Además de las características de centralidad, se investigó el tejido en el que dichos genes se encontraban sobreexpresados con fines descriptivos. Para esto, usamos Human Protein Atlas (HPA), 27 una base de datos desarrollada por el Instituto Karolinska en asociación con otras instituciones, en la que se centraliza información de expresión de transcriptos proveniente de diferentes fuentes. Agregado a esto, paquetes adicionales de Cytoscape fueron instalados para detectar regiones densas o clúster de proteínas. La aplicación Molecular complex detection (Mcode) 28 fue utilizada para este fin, el análisis se llevó a cabo con un Degree de 3, Node score 0,2 y un K-score de 2.
Análisis funcional
El análisis de enriquecimiento funcional de los genes se llevó a cabo usando g.Profiler (https://biit.cs.ut.ee/gprofiler/gost). 29 Este servidor público otorga una colección de herramientas usadas de manera estandarizada en análisis biológicos, lo que permite la identificación de vías biológicas y categorías de ontología genética, que incluyen procesos biológicos (BP: biological processes), funciones moleculares (MF: molecular functions) y componentes celulares (CC: celular components). El método de corrección de Benjamini-Hochberg fue utilizado para el cálculo del false discovery rate (FDR). Se establecieron valores de p < 0,001 y de FDR < 0,05 como significativos. Adicionalmente las anotaciones electrónicas automáticas fueron excluidas y al menos 5 términos query fueron requeridos para aceptar una anotación funcional, con el fin de reducir la probabilidad de falsos positivos. A continuación, fueron seleccionaron los términos de ontología más significativos según el FDR y, usando la herramienta REVIGO (Reduce and Visualize Gene Ontology, http:// revigo.irb.hr/), 30 se removieron los términos redundantes.
Consideraciones éticas
No aplicable
RESULTADOS
Lista de genes
Se obtuvieron 5605, 759, 2787 y 532 genes relacionados con los fenotipos de obesidad, HTA, SM y RCIU, respectivamente. Al aplicar teoría de conjuntos se encontraron 343 genes superpuestos entre la RCIU y las otras 3 entidades (Figura 1).
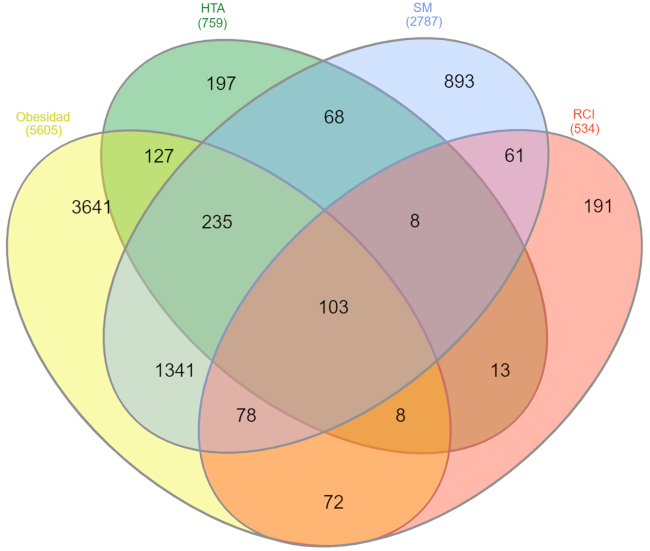 Fig. 1
Diagrama de Venn de genes alterados en los fenotipos de obesidad, hipertensión arterial, síndrome metabólico y restricción del crecimiento intrauterino
Fig. 1
Diagrama de Venn de genes alterados en los fenotipos de obesidad, hipertensión arterial, síndrome metabólico y restricción del crecimiento intrauterino
Red de interacción genética
Los 343 genes resultantes de la superposición se utilizaron para la creación de las redes. La red obtenida contenía 12633 conexiones (Figura 2).
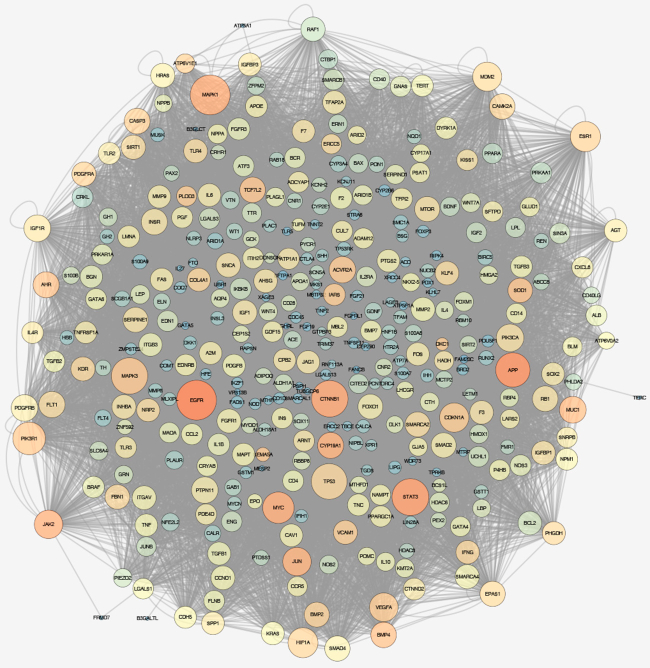 Fig. 2
Red de integración genética unificada de los 343 nodos con 12633 interacciones. El diámetro del nodo se relaciona directamente con el grado de centralidad y el color con la intermediación, siendo mayor el valor para ese gen cuanto más similar es al color rojo.
Fig. 2
Red de integración genética unificada de los 343 nodos con 12633 interacciones. El diámetro del nodo se relaciona directamente con el grado de centralidad y el color con la intermediación, siendo mayor el valor para ese gen cuanto más similar es al color rojo.
Subsecuentemente, se obtuvo el top 20 de genes: EGFR, receptor del factor de crecimiento epidérmico; MAPK1, proteína quinasas activadas por mitógenos 1; STAT3, transductor de señal y activador de la transcripción 3, CTNNB1, beta-catenina 1; MAPK3, proteína quinasas activadas por mitógenos 3; TP53, proteína supresora de tumores 53; APP, proteína precursora amiloidea; PIK3R1, subunidad reguladora 1 de fosfoinositol 3-quinasas; MYC, protooncogen bHLHe39, proteína básica hélice-bucle-hélice 39; ESR1, receptor de estrógeno alfa; HIF1A, subunidad alfa del factor 1 inducible por hipoxia; CDKN1A, inhibidor de la quinasa dependiente de ciclina 1A; MDM2, protooncogen MDM2; VEGFA, factor de crecimiento endotelial vascular A; JAK2, janus quinasa 2; JUN, C-jun, subunidad factor de transcipción AP-1; PIK3CA, subunidad 1 alfa catalítica de fosfoinositol-3-quinasas; IGF1R, receptor del factor de crecimiento similar a la insulina 1; MUC1, mucina epitelial 1; PDGFRB, factor de crecimiento derivado de plaquetas. Seleccionados según los resultados del análisis topológico, los valores resultantes fueron de 128 a 239 y 0,00342065 a 0,0131197 para el grado de centralidad e intermediación, respectivamente (Tabla 1). En la misma tabla, se muestran los términos de ontología genética, la función principal del gen y el tejido donde es sobreexpresado. Como puede observase en la tabla, algunos de estos genes tienen una expresión ubicua (Tabla 1).
Tabla 1
Top 20 de genes centrales de la red de interacción
 Términos GO: Términos de ontología genética; STAT: transductor de señales y activador de la transcripción; VEGFA: Factor de crecimiento endotelial vascular A; PDGF: Factor de crecimiento derivado de plaquetas; PI3K: Fosfoinositol 3-quinasa; AKT1: Gen que codifica para proteina quinasa B; PKB: Proteína quinasa B; MAPK: Proteína quinasa activada por mitógenos; ERK: Proteína quinasa regulada por señales extracelulares; SRC: Proteína quinasa SRC; NFkappa-B: Factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas. Con * se identifican los genes de distribución ubicua.
Términos GO: Términos de ontología genética; STAT: transductor de señales y activador de la transcripción; VEGFA: Factor de crecimiento endotelial vascular A; PDGF: Factor de crecimiento derivado de plaquetas; PI3K: Fosfoinositol 3-quinasa; AKT1: Gen que codifica para proteina quinasa B; PKB: Proteína quinasa B; MAPK: Proteína quinasa activada por mitógenos; ERK: Proteína quinasa regulada por señales extracelulares; SRC: Proteína quinasa SRC; NFkappa-B: Factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas. Con * se identifican los genes de distribución ubicua.
Clústeres de proteínas
Luego de haber identificado los genes codificantes de proteínas más importantes de la red, se realizó un análisis más amplio de la red. Como se ha mencionado previamente, la red está conformada por genes superpuestos entre la RCIU y patologías que representan un riesgo cardiovascular como obesidad, HTA y SM. Por lo tanto, se procedió a localizar zonas densas de complejos de proteínas para, luego, continuar con la identificación de términos de ontología implicados en cada uno de los clústeres hallados (Figura 3). Se excluyeron los clústeres conformados por menos de 3 nodos, debido a que podrían mostrar resultados poco confiables en la determinación de términos de ontología. Como resultado, ocho clús-teres fueron identificados, conformados por entre 5 y 50 nodos (Figura 3).
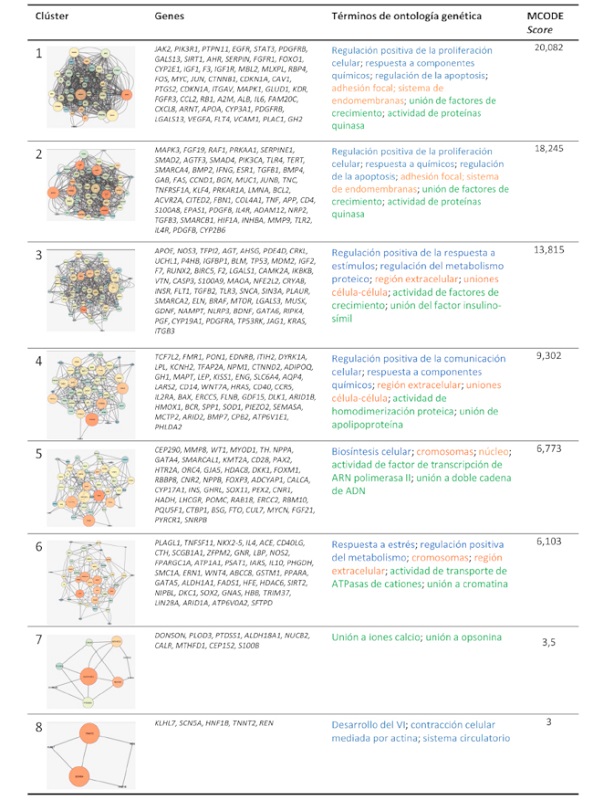 Fig. 3
Clústeres de proteínas y términos de ontología más significativos identificados. En cuanto a los términos de ontología, en color azul se identifican aquellos que corresponden a los procesos biológicos, en color naranja a aquellos que corresponden a los compartimientos celulares, y en color verde a aquellos que corresponden a la función molecular
Fig. 3
Clústeres de proteínas y términos de ontología más significativos identificados. En cuanto a los términos de ontología, en color azul se identifican aquellos que corresponden a los procesos biológicos, en color naranja a aquellos que corresponden a los compartimientos celulares, y en color verde a aquellos que corresponden a la función molecular
DISCUSIÓN
Este estudio es el primero, según nuestro conocimiento, en utilizar el análisis de redes para evaluar la interacción compleja entre la RCIU y los fenotipos de obesidad, HTA, y SM, en búsqueda de genes cardinales que tendrían potencial función como biomarcadores putativos del desarrollo de estos fenotipos en individuos que presentaron RCIU o que podrían ser empleados como potenciales blancos terapéuticos.
Identificamos un total de 20 genes que resultaron significativamente relevantes en el análisis de la red de interacción, la gran mayoría de estos genes estuvieron relacionados con vías de señalización intracelular asociadas a respuesta a sustancias orgánicas, y a regulación positiva de la biosíntesis celular. De acuerdo con los resultados obtenidos, al menos cuatro de estos genes identificados codifican para factores de crecimiento o sus receptores, VEGFA, PDGFRB, EGFR e IGF1R. Es de destacar la identificación de IGF1R, cuyo ligando, el factor de crecimiento similar a la insulina 1 (IGF1), es sintetizado en el hígado, aunque se expresa ubicuamente, y puede modular el metabolismo de hidratos de carbono y lípidos. Su liberación es estimulada por la hormona de crecimiento y la insulina. Cuando hay restricción calórica, los niveles de IGF-1 disminuyen y su síntesis en el hígado es refractaria a la estimulación de la hormona de crecimiento. 31 Este proceso limita el crecimiento y la síntesis de proteínas cuando la disponibilidad de nutrientes se ve comprometida. Por otra parte, en estados fisiopatológicos, incluido el aumento de la resistencia a la insulina, el número de receptores IGF1R cambia, lo que anula la posibilidad de que el IGF-1 module el metabolismo de la glucosa, por lo que se lo asocia al desarrollo o mantenimiento del SM. 32
Por otra parte, la identificación de cuatro genes cardinales que se expresan en tejidos estrechamente relacionados con los fenotipos estudiados, como la placenta, los distintos tipos de músculo y el tejido adiposo, resultan de interés, ya que aún no han sido evaluados en la práctica como biomarcadores en los fenotipos incluidos en este estudio.
El gen CTNNB1 está involucrado en la internalización de la insulina y la expresión de su proteína se ha encontrado como vía de señalización involucrada en la hipertrofia cardíaca, 33 además de modular negativamente la diferenciación de adipocitos. 34 Por lo tanto, este gen puede ser de interés para los tres fenotipos estudiados.
El gen APP, que se expresa en la placenta y en el tejido muscular cardíaco y liso, de acuerdo con el análisis de HPA, está relacionado con los procesos de transcripción celular. Su identificación resulta de interés, ya que comúnmente se lo asocia con la enfermedad de Alzheimer, pero actualmente se asocia también al SM. La vía de señalización intracelular de la insulina está involucrada en el metabolismo de la proteína amiloide, a la vez que esta puede unirse a los receptores de insulina, lo que desencadena su internalización y disminuye así la respuesta de las neuronas a la insulina y promueve la resistencia a la insulina. 35
Además, la identificación de dos protooncogenes, MYC y MDMD2, en el análisis de redes de interacción génica pone de manifiesto la relevancia de los procesos de progresión del ciclo celular y la diferenciación celular. Ambos con funciones relacionadas con el mantenimiento de la homeostasis cardiovascular, han sido escasamente explorados en la bibliografía en relación con estos fenotipos, y su inclusión como posibles biomarcardores o blancos terapéuticos también resulta de interés.
Por otra parte, el análisis de clústeres nos permitió conocer los términos de ontología genética más significativos, que se clasificaron según el valor del FDR o p corregido, por lo que eran más significativos cuanto menor era el valor obtenido. Entre los términos ontológicos, se destacan aquellos relacionados con los procesos biológicos de proliferación y muerte celular programada, de comunicación intercelular, del metabolismo proteico y de desarrollo del sistema cardiovascular. Estos hallazgos son coincidentes con la evidencia que muestra estos procesos como claves del desarrollo de alteraciones en órganos y tejidos. 9,36 También hallamos que los genes identificados codifican proteínas que pueden hallarse en el compartimiento extracelular, lo que permitiría su utilización efectiva como biomarcadores, y que sus funciones moleculares están estrechamente relacionadas con factores de crecimiento y con estabilidad de ADN.
La principal fortaleza de este estudio fue el aprovechamiento de la información globalmente disponible, mediante el procesamiento y el análisis de los datos de diferentes niveles de información biológica. Además, empleamos métodos con análisis estadísticos con un alto grado de confiabilidad. Por otra parte, este análisis permite orientar estudios posteriores; esto reduce el tiempo y los recursos necesarios para la evaluación de biomarcadores y objetivos terapéuticos, lo que permite un abordaje mejor dirigido.
Sin embargo, existen ciertas limitaciones, ya que puede presentarse variabilidad poblacional en los resultados obtenidos, y es necesaria la evaluación experimental que corrobore nuestros hallazgos. A pesar de esto, la utilización de seis bases de datos para obtener inicialmente el conjunto de genes de cada fenotipo, sumado al uso de tres herramientas de distinta metodología para establecer las redes de interacción genética, asegura complementariedad entre ellas.
Por lo tanto, podemos concluir que los genes hallados en este estudio podrían ser de utilidad como biomarcadores putativos de la presencia de alteraciones cardiovasculares y metabólicas asociadas con la RCIU, o podrían ser de utilidad como potenciales blancos terapéuticos de estrategias de tratamiento orientadas al genotipo del paciente.
BIBLIOGRAFÍA
Armengaud JB, Yzydorczyk C, Siddeek B, Peyter AC, Simeoni U. Intrauterine growth restriction: clinical consequences on health and disease at adulthood. Reprod Toxicol 2020; 99:168-76. doi: 10.1016/j.reprotox.2020.10.005.
ACOG Practice Bulletin No. 204. Obstet Gynecol 2019;133: e97-e109. doi:10.1097/aog.0000000000003070
Fescina RH, De Mucio B, Martínez G, Alemán A, Sosa C, Mainero L, Rubino M. Vigilancia del crecimiento fetal: manual de autoinstrucción. 2 ed. Montevideo: CLAP/SMR; 2013. (CLAP/SMR. Publicación científica; 1586).
Unterscheider J, Daly S, Geary MP, Dicker P, Tully EC, Malone FD. Optimizing the definition of intrauterine growth restriction: the multicenter prospective PORTO Study. Res Obstetr 2013; 208:290.e1-290.e6. doi: 10.1016/j.ajog.2013.02.007
Menéndez-Castro C, Rascher W, Hartner A. Intrauterine growth restriction - impact on cardiovascular diseases later in life. Mol Cell Pediatr 2018;5:4. doi: 10.1186/s40348-018-0082-5.
Crispi F, Miranda J, Gratacos E. Long-term cardiovascular consequences of fetal growth restriction: biology, clinical implications, and opportunities for prevention of adult disease. Am J Obstet Gynecol 2018;218(2S):S869-S79. doi: 10.1016/j.ajog.2017.12.012.
Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986;1(8489):1077-81. doi: 10.1016/s0140-6736(86)91340-1.
Barker DJ. The developmental origins of adult disease. J Am Coll Nutr 2004;23(6 Suppl):588S-95S. doi: 10.1080/07315724.2004.10719428.
Tomat AL, Veiras LC, Aguirre S, Fasoli H, Elesgaray R, Caniffi C, et al. Mild zinc deficiency in male and female rats: early postnatal alterations in renal nitric oxide system and morphology. Nutrition 2013;29:568-73. doi: 10.1016/j.nut.2012.09.008.
Nyrnes SA, Garnæs KK, Salvesen Ø, Timilsina AS, Moholdt T, Ingul CB. Cardiac function in newborns of obese women and the effect of exercise during pregnancy. A randomized controlled trial. PLoS One 2018;13:e0197334. doi: 10.1371/journal.pone.0197334.
Van De Maele K, Devlieger R, Gies I. In utero programming and early detection of cardiovascular disease in the offspring of mothers with obesity. Atherosclerosis 2018;275:182-95. doi: 10.1016/j.athero-sclerosis.2018.06.016.
Devaskar SU, Chu A. Intrauterine Growth Restriction: Hungry for an Answer. Physiology (Bethesda) 2016;31:131-46. doi: 10.1152/physiol.00033.2015.
Fall CHD. Fetal programming and the risk of noncommunicable disease. Indian J Pediatr 2013;80 Suppl 1:S13-20. doi: 10.1007/s12098-012-0834-5.
Briffa JF, Wlodek ME, Moritz KM. Transgenerational programming of nephron deficits and hypertension. Semin Cell Dev Biol 2020;103:94-103. doi: 10.1016/j.semcdb.2018.05.025.
Cuarta Encuesta Nacional de Factores de Riesgo para Enfermedades no Transmisibles. Ministerio de Salud y Desarrollo Social de la Nación Argentina. 2019.
Amihaeseiˇ IC, Chelaru L. Metabolic syndrome a widespread threatening condition; risk factors, diagnostic criteria, therapeutic options, prevention and controversies: an overview. Rev Med Chir Soc Med Nat Iasi 2014;118:896-900.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46:D1062-7, doi:10.1093/ nar/gkx1153
Pinero J, Queralt-Rosinach N, Bravo À, Deu-Pons J, Bauer-Mehren A, Baron M, Sanz F, et al. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford) 2015;2015:bav028. doi:10.1093/database/bav028
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019:47: D1005-12, doi:10.1093/nar/gky1120
Mattingly CJ, Colby GT, Forrest JN, Boyer JL. The Comparative Toxicogenomics Database (CTD). Environ Health Perspect 2003;111:793-5, doi:10.1289/ehp.6028
Szklarczyk, D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447-52, doi:10.1093/nar/gku1003
Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R, et al. Consensus PathDB: toward a more complete picture of cell biology. Nucleic Acids Res 2011;39:D712-7, doi:10.1093/nar/gkq1156
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 2010;38:W214-20, doi:10.1093/nar/gkq537
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498-504, doi:10.1101/gr.1239303.
Pavlopoulos GA, Secrier M, Moschopoulos CN, Soldatos TG, Kossida S, Aerts J, et al. Using graph theory to analyze biological networks. BioData Min 2011;4:10. doi:10.1186/1756-0381-4-10.
Xiong W, Xie L, Zhou S, Liu H, Guan J. The centrality of cancer proteins in human protein-protein interaction network: a revisit. Int J Comput Biol Drug Des 2014;7:146-56. doi:10.1504/ IJCBDD.2014.061643
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419, doi:10.1126/science.1260419.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003;4:2. doi:10.1186/1471-2105-4-2.
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019;47:W191-W198, doi:10.1093/nar/gkz369.
Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS One 2011; 6:e21800. doi:10.1371/journal.pone.0021800
Clemmons DR. Involvement of insulin-like growth factor-I in the control of glucose homeostasis. Curr Opin Pharmacol 2006;6:620-5. doi: 10.1016/j.coph.2006.08.006.
Aguirre GA, Rodríguez De Ita J, de la Garza RG, Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J Transl Med. 2016;14:3. doi: 10.1186/s12967-015-0762-z.
Hoogeboom D, Burgering BM. Should I stay or should I go: beta-catenin decides under stress. Biochim Biophys Acta. 2009;1796:63-74. doi: 10.1016/j.bbcan.2009.02.002.
Lee SH, Kim B, Oh MJ, Yoon J, Kim HY, Lee KJ, Lee JD, Choi KY. Persicaria hydropiper (L.) Spach and its Flavonoid Components, Isoquercitrin and Isorhamnetin, Activate the Wnt/β-catenin Pathway and Inhibit Adipocyte Differentiation of 3T3-L1 Cells. Phytother Res 2011;25:1629-35. doi: 10.1002/ptr.3469
Campos-Peña V, Toral-Ríos D, Becerril-Pérez F, Sánchez-Torres C, Delgado-Namorado Y, Torres-Ossorio E, et al. Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: Is Aβ a Crucial Factor in Both Pathologies? Antioxid Redox Sign 2017;26:542-60. doi: 10.1089/ars.2016.6768.
Cañas D, Herrera EA, García-Herrera C, Celentano D, Krause BJ. Fetal Growth Restriction Induces Heterogeneous Effects on Vascular Biomechanical and Functional Properties in Guinea Pigs (Cavia porcellus). Front Physiol 2017;8:144. doi: 10.3389/fphys.2017.00144.
Notas
Notas
Financiamiento Este trabajo fue financiado por la Universidad de Buenos Aires (UBA) [UBACYT 20020170200093BA, 2018-2019; 20020190200227BA, 2020-2021]; y el Instituto de Química y Metabolismo del Fármaco (IQUIMEFA), UBA-CONICET, Argentina.
Declaración de intereses
Conflicto de interés Los autores declaran que no poseen conflictos de intereses. (Véase formulario de conflicto de intereses de los autores en la web / Material suplementario).
Notas de autor
Criterios de autoría IDAM: Obtención, análisis e interpretación de datos, redacción del artículo; DAS, AT, RE, CA: Interpretación de datos y revisión del artículo; CC: Diseño, interpretación de datos, redacción y revisión del artículo.
Dirección para separatas: Carolina Caniffi - Cátedra de Fisiología - Junín 956, piso 7, CABA (C1113AAD), Argentina - Teléfono: +541152874713 - E-mail: ccaniffi@ffyb.uba.ar