INTRODUCCIÓN
El SQTL congénito se caracteriza por la prolongación del potencial de acción del miocardio ventricular debido al aumento de las corrientes de entrada de sodio y calcio (INa e ICaL ) o la disminución de las corrientes de salida de potasio (IKs, IKr e IK1). Hasta el momento se identificaron mutaciones en 20 genes diferentes que codifican los canales iónicos cardíacos y/o proteínas moduladoras que intervienen de manera directa o indirecta en la formación de estas corrientes. (1)
Con una prevalencia en la población general de 1 en 2000 individuos, ya no es una situación excepcional ver a un paciente con un intervalo QT prolongado en una consulta médica cotidiana para práctica de deporte, escolaridad o prelaboral. (2)
Con el avance de la tecnología informática e ingeniería genética, se han identificado centenares de variantes en cada uno de los genes involucrados en el SQTL que, a través de diferentes tipos de mutaciones alteran el funcionamiento de los canales iónicos de los miocitos. La mayoría de las mutaciones muestran un patrón de herencia autosómico dominante. De esta manera, los individuos nacen con la mutación causal de la enfermedad y conviven con ella a lo largo de toda su vida.
Todos los individuos afectados presentan una manifestación fenotípica en común, la prolongación de la duración de la repolarización ventricular. Sin embargo, la existencia de una amplia variabilidad clínica entre los pacientes plantea interrogantes sobre los factores epigenéticos que modulan su expresión fenotípica. Aun cuando los pacientes nacen con el genotipo patogénico, el fenotipo puede no manifestarse nunca, evidenciarse tardíamente o en algunos casos, sólo hacerlo de manera intermitente, expresándose sólo en determinados días y pudiendo permanecer por periodos prolongados de tiempo totalmente asintomáticos. Así mismo, el riesgo de padecer eventos cardíacos adversos puede variar entre los portadores de la misma variante genética y aún en la misma persona, dependiendo de la situación a la que fue expuesta. (3)
En este trabajo, hemos evaluado las características clínicas y genéticas de nuestros pacientes con diagnóstico de SQTL mediante un estudio genético con secuenciación masiva en paralelo y un seguimiento clínico a largo plazo por más de 10 años, pudiendo así valorar, tanto la historia natural de la enfermedad, como la aparición de eventos cardiacos adversos (síncope, taquiarritmia ventricular y/o muerte súbita cardíaca), la respuesta al tratamiento farmacológico y/o dispositivos antiarrítmicos implantables, y las complicaciones asociadas a ellos.
MATERIAL Y MÉTODOS
Se diseñó un estudio retrospectivo seleccionando a los individuos con sospecha de síndrome QT largo congénito que concurren al Hospital General de Ramos Mejía. Se utilizaron los siguientes criterios de inclusión: hombres y mujeres con edad de 5 a 70 años; sospecha clínica de SQTL congénito (un intervalo QTc ≥480 milisegundos en el ECG o puntuación de Schwartz ≥3); identificación de una variante patogénica de SQTL congénito en estudio genético. Se excluyó a aquellos con alguno de los siguientes criterios: SQTL adquirido por medicamentos; negación a firmar el consentimiento informado; menores de 18 años, sin consentimiento de los progenitores o tutores legales; hallazgo negativo de estudio genético. De cada uno de ellos, se obtuvieron registros de ECG de 12 derivaciones simultáneas. Los intervalos QTc fueron corregidos en función de la frecuencia cardíaca mediante la fórmula de Bazett (QTc = QT/√RR, en segundos).
Para el análisis de secuencias y las pruebas de deleción/duplicación se utilizó el método de secuenciación masiva en paralelo (NGS) con un panel de más de 150 genes para arritmias y miocardiopatías (ABCC9, ACADVL, ACTC1, ACTN2, AGL, ALMS1, ALPK3, BAG3, BRAF, CACNA1C, CACNA1D, CALM1, CALM2, CALM3, CASQ2, CBL, CDH2, CPT2, CRYAB, CSRP3, DES, DMD, DNAJC19, DOLK, DSC2, DSG2, DSP, ELAC2, EMD, EYA4, FHL1, FKRP, FKTN, FLNC, GAA, GATA4, GATA5, GJA5, GLA, HCN4, HRAS, JUP, KCNE1, KCNH2, KCNJ2, KCNQ1, KRAS, LAMP2, LMNA, LZTR1, MAP2K1, MAP2K2, MRAS, MTO1, MYBPC3, MYH7, MYL2, MYL3, MYL4, MYLK3, NF1, NKX2-5, NRAS, PCCA, PCCB, PKP2, PLN, PPA2, PPCS, PPP1CB, PRKAG2, PTPN11, RAF1, RASA1, RBM20, RIT1, RYR2, SCN5A, SDHA, SGCD, SHOC2, SLC22A5, SOS1, SOS2, SPRED1, TAZ, TBX20, TCAP, TMEM43, TMEM70, TNNC1, TNNI3, TNNI3K, TNNT2, TPM1, TRDN, TRPM4, TTN, TTR, VCL, A2ML1, AKAP9, ANK2, ANKRD1, CACNA2D1, CACNB2, CALR3, CAV3, CHRM2, CTF1, CTNNA3, DTNA, FHL2, GATA6, GATAD1, GPD1L, HAND1, ILK, JPH2, KCNA5, KCND3, KCNE2, KCNE3, KCNE5, KCNJ5, KCNJ8, KCNK3, KIF20A, KLF10, LAMA4, LDB3, LRRC10, MAP3K8, MED12, MYH6, MYLK2, MYOM1, MYOZ2, MYPN, NEBL, NEXN, NPPA, PDLIM3, PLEKHM2, PRDM16, RANGRF, RASA2, RRAS, SCN10A, SCN1B, SCN2B, SCN3B, SCN4B, SLMAP, SNTA1, TMPO, TXNRD2), desarrollado por Invitae® (1400 16th Street, San Francisco, CA 94103, EE. UU.). El ADN genómico obtenido de la muestra de sangre periférica se enriqueció para las regiones objetivo mediante un protocolo basado en hibridación y se secuenció con tecnología Illumina®. Todas las regiones objetivo se secuenciaron con una profundidad ≥50x o se complementaron con análisis adicionales. Las lecturas se alinearon con una secuencia de referencia (GRCh37) y los cambios de secuencia se identificaron e interpretaron en el contexto de una única transcripción clínicamente relevante. El enriquecimiento y el análisis se centraron en la secuencia codificante de las transcripciones indicadas, 20 pb de la secuencia intrónica flanqueante y otras regiones genómicas específicas que se demostró que eran causantes de la enfermedad en el momento del diseño del ensayo. Las variantes detectadas se evaluaron consultando las siguientes bases de datos: dbSNP de NCBI (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/snp/), ExAC (Exome Aggregation Consortium, https://exac.broadinstitute.org), gnomAD (Genome Aggregation Database, https://gnomad.broadinstitute.org/) y OMIM (Online Mendelian Inheritance in Man, https://omim.org/). La patogenicidad de las variantes fue estimada mediante tres tipos diferentes de software de predicción: SIFT (https://sift.bii.a-star.edu.sg/), PolyPhen-2 (https://genetics.bwh.harvard.edu/pph2/) y Align-GVGD (https://bio.tools/align-gvgd/).
Las variantes se clasificaron como patogénica o probablemente patogénica, de significado incierto, probablemente benigna o benigna utilizando un sistema de puntuación de evidencia basado en el consenso del Colegio Americano de Genética Médica y Genómica y la Asociación de Patología Molecular. (4) Los resultados se clasificaron como positivos, negativos, portadores o inciertos según la clasificación de la variante identificada y el patrón de herencia de la afección asociada.
Análisis estadístico
Las variables cuantitativas fueron expresadas en promedio ± desviación estándar (DE). Las variables cualitativas fueron expresadas como porcentajes (%).
Consideraciones éticas
El estudio se realizó de acuerdo con las normas éticas de la Declaración de Helsinki, y el protocolo del estudio fue aprobado por el Comité de Ética del Hospital General de Agudos Dr. José María Ramos Mejía, Ciudad Autónoma de Buenos Aires, Argentina.
DISCUSIÓN
El SQTL congénito descrito por Jervell y Lange-Nielsen en 1957 y por Romano y Ward en 1964 es una canalopatía hereditaria que se caracteriza por una alteración en la repolarización ventricular y se manifiesta por una prolongación anormal en la duración de intervalo QTc. Predispone a la aparición de las taquiarritmias ventriculares potencialmente letales (torsade de pointes y/o fibrilación ventricular) que suele ocurrir por el aumento del tono adrenérgico tras los estímulos auditivos, el ejercicio físico o estrés emocional. (5,6)
En los últimos 25 años, 20 genes fueron asociados con el SQTL congénito. Sin embargo, un análisis reciente clasificó nuevamente a varios de estos genes como de evidencia limitada o controvertida. (7) Este enfoque dejó siete genes con evidencia definitiva o sólida de causalidad (KCNQ1, KCNH2, SCN5A, CALM1, CALM2, CALM3 y TRDN). Todos estos genes codifican canales iónicos involucrados en la repolarización cardíaca o proteínas que regulan o modulan la función del canal iónico.
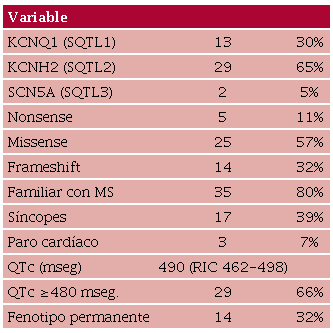
El 90% de los individuos con genotipo de SQTL son portadores de mutaciones en uno de los 3 genes principales de la enfermedad: el KCNQ1 (SQTL tipo 1), el KCNH2 (SQTL tipo 2) y el SCN5A (SQTL tipo 3), que codifican a las subunidades alfa de los canales iónicos Kv7.1 (IKs), Kv11.1 (IKr) y Nav1.5 (INa), respectivamente (Figura 1). (8,9)
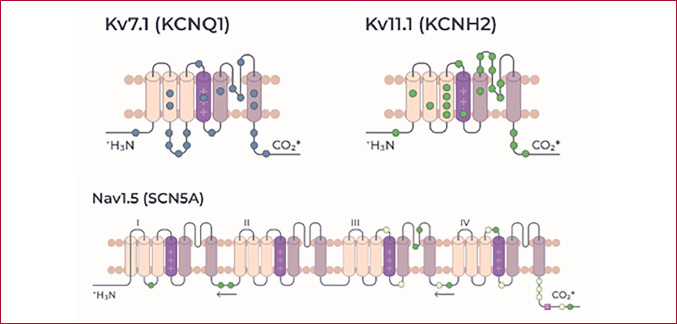 Figura 1
Canales iónicos involucrados en SQTL tipo 1, tipo 2 y tipo 3. El gen KCNQ1 codifica a la subunidad alfa del canal de potasio voltaje dependiente Kv7.1 (responsable de corriente rectificadora IKs). El gen KCNH2 codifica a la subunidad alfa del canal de potasio voltaje dependiente Kv11.1 (responsable de corriente rectificadora IKr). El canal de sodio voltaje dependiente Nav1.5 (responsable de corriente INa) es codificado por gen SCN5A.
Figura 1
Canales iónicos involucrados en SQTL tipo 1, tipo 2 y tipo 3. El gen KCNQ1 codifica a la subunidad alfa del canal de potasio voltaje dependiente Kv7.1 (responsable de corriente rectificadora IKs). El gen KCNH2 codifica a la subunidad alfa del canal de potasio voltaje dependiente Kv11.1 (responsable de corriente rectificadora IKr). El canal de sodio voltaje dependiente Nav1.5 (responsable de corriente INa) es codificado por gen SCN5A.
Aproximadamente el 40% de las mutaciones corresponden a mutaciones sin sentido (nonsense), que consisten en una mutación puntual en una secuencia de ADN que da como resultado un codón de terminación prematura), o a mutaciones de cambio de marco de lectura (frameshift) causadas por la inserción o deleción de nucleótidos en una secuencia de ADN, lo que genera un marco de lectura completamente diferente del original. Estas mutaciones alteran la síntesis proteica y generan subunidades alfa defectuosas de los canales iónicos. El 60% restante, son mutaciones con cambio de sentido, de sentido erróneo o contrasentido (missense), donde un solo cambio de nucleótido altera un codón de un aminoácido (Figura 2). Estas mutaciones pueden alterar la permeabilidad del poro, la activación/desactivación o el tráfico intracelular de los canales iónicos. (10)
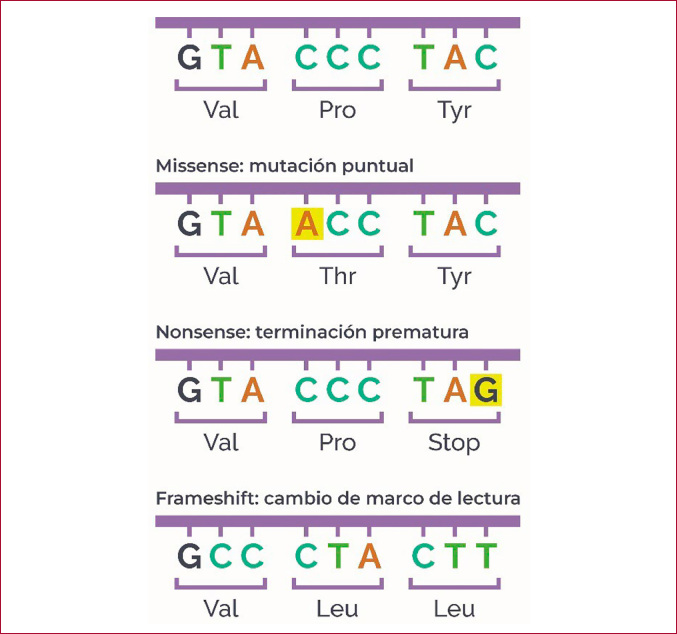 Figura 2
Ejemplo de tipos de mutaciones. Missense: mutación puntual de ADN que cambia un aminoácido. Nonsense: mutación puntual de ADN que introduce un codón de terminación prematura. Frameshift: inserción o deleción de ADN con cambio de marco de lectura. Modificada de: Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization Physiol Rev 2005;85:1205-53. https://10.1152/physrev.00002.2005
Figura 2
Ejemplo de tipos de mutaciones. Missense: mutación puntual de ADN que cambia un aminoácido. Nonsense: mutación puntual de ADN que introduce un codón de terminación prematura. Frameshift: inserción o deleción de ADN con cambio de marco de lectura. Modificada de: Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization Physiol Rev 2005;85:1205-53. https://10.1152/physrev.00002.2005
Secuencia de ADN: A: adenina; T: timina; C: citosina; G: guanina. Secuencia de aminoácidos: Ser: serina; Val: valina; Pro: prolina; Tyr: tirosina; Thr: treonina; Leu: leucina; Stop: codón de terminación prematura.
En nuestro estudio, los pacientes mostraron un perfil genético similar a los estudios mencionados. El 95% de los pacientes eran portadores del genotipo SQTL tipo 1 (KCNQ1) o de SQTL tipo 2 (KCNH2). Las variantes genéticas predominantes se debieron a mutaciones puntuales missense con cambio de un sólo aminoácido en la secuencia proteica.
Con frecuencia, el carácter intermitente de la manifestación del fenotipo de SQTL dificulta el diagnóstico de los pacientes. En nuestro estudio, la mayoría de los pacientes (66%) manifestó el fenotipo de SQTL (QTc >480 mseg) pero sólo un tercio tuvo el fenotipo permanente. Estos resultados fueron comparables a los resultados del estudio de Yoo et al., en el cual los pacientes con SQTL mostraron oscilaciones significativas del intervalo QT en diferentes mediciones realizadas y sólo el 20% mantuvo el fenotipo permanente. (11) Estos hallazgos sugieren que para el diagnóstico de SQTL congénito, los pacientes deben ser evaluados de manera minuciosa y contínua mediante ECG seriados, ECG de esfuerzo o registro dinámico con monitoreo Holter de manera periódica. En estas circunstancias, el estudio genético para identificar la variante patogénica responsable se torna crucial para detectar a los pacientes con SQTL congénito de manera precisa y precoz.
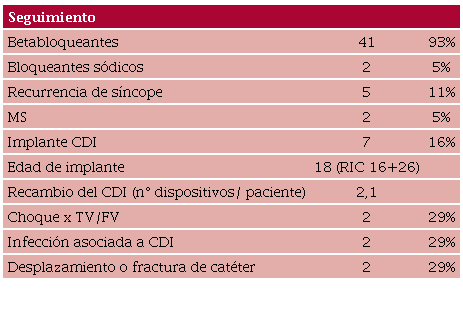
A menudo, los individuos con SQTL suelen manifestar el fenotipo y padecer episodios sincopales y/o muerte súbita a edades tempranas. La mortalidad de los pacientes con SQTL oscila entre 1 a 2% en 5 años. (12) Los betabloqueantes son efectivos, especialmente en el SQTL1 en cual la TV/FV es gatillada por el esfuerzo. El incumplimiento del tratamiento y el uso de fármacos que prolongan el intervalo QT son responsables principales de fracasos terapéuticos. (13,14) En el SQTL2 y el SQTL3, los eventos arrítmicos letales suelen desencadenarse en reposo o ante estímulo auditivo o emocional. (15,16) Entre los individuos con un cardiodesfibrilador implantable (CDI), la tasa de eventos recurrentes es de aproximadamente el 3% a 28% en 5 años. (17,18)
Existe aún la tendencia a considerar innecesaria la identificación del genotipo de los pacientes con SQTL, una vez efectuado el diagnóstico mediante los criterios clínicos. Esta conducta imposibilita el inicio del cribado en cascada de la familia afectada. Considerando que la respuesta a los fármacos (betabloqueantes vs bloqueantes sódicos) y los estímulos que actúan como gatillo arritmogénico (esfuerzo, auditivo o emocional) son muy diferentes entre los genotipos, el desconocimiento de la causa genética dificulta brindar una terapia adecuada a los pacientes. Las consecuencias podrían resultar en la ocurrencia de muertes que podrían evitarse, en especial, entre los individuos con genotipo positivo y fenotipo negativo. De esta manera, la biología molecular ya no debe considerarse como campo exclusivo de investigación sino una herramienta médica esencial y cotidiana. (19)
En nuestro estudio, el implante de CDI fue indicado entre la segunda y tercera década de la vida. El motivo principal fue la ocurrencia de síncopes recurrentes aún con terapia betabloqueante (prevención primaria). Las 2 mujeres (una con SQTL1 y otra SQTL2) que recibieron un CDI para prevención secundaria tuvieron terapias adecuadas por TV/FV luego de transcurrir 2 a 6 años del implante. Por otro lado, la tasa de complicaciones asociadas a los dispositivos (infección o desplazamiento/fractura de catéter durante el seguimiento a largo plazo) fue elevada. Según las guías vigentes, el implante de CDI está indicado para fines de prevención secundaria (en individuos que padecieron un paro cardíaco reanimado, clase I) y primaria (en aquellos con síncopes recurrentes bajo tratamiento betabloqueante, clase IIa). (20) Evidentemente, la decisión de implantar el CDI salva la vida de los pacientes de alto riesgo de MS cardíaca. Sin embargo, en el SQTL congénito, al igual que en otras canalopatías hereditarias, la edad muy temprana de diagnóstico e implante de los dispositivos y la elevada tasa (más de 20% en 5 a 10 años) de complicaciones asociadas (infección, perforación miocárdica, desplazamiento, desgaste y/o fractura del catéter, consecuencias psicológicas, etc) que ocurren a lo largo de la vida de los pacientes, sugieren que la decisión de indicar el CDI sea basada en una evaluación minuciosa y cautelosa. (21,22)
 PAOLA SETTEPASSI paosettepassi@gmail.com
PAOLA SETTEPASSI paosettepassi@gmail.com