CARTA CIENTÍFICA
Las investigaciones sobre la predisposición hereditaria a la hipertensión arterial pulmonar (HAP) han conducido a la identificación de variantes en el gen que codifica el receptor de la proteína morfogenética ósea tipo II (BMPR2). Alrededor del 70 al 80 % de los casos con HAP hereditaria (HAPH) y hasta un 40 % de los casos idiopáticos (HAPI) son causados por variantes genéticas en BMPR2. (1)
Han sido descriptas más de 800 variantes diferentes del mismo; sin embargo, la penetrancia es reducida, ya que solo del 20 al 30 % de los portadores devienen en HAP. Esto último sugiere la contribución de otros factores genéticos, epigenéticos, ambientales y hormonales en la modulación y desarrollo de la enfermedad. (2,3,4)
BMPR2 forma parte de una de las dos grandes vías de señalización que conforman la superfamilia del factor de crecimiento transformante-β (TGF-β): la vía de la proteína morfogenética ósea (BMP)-factor de diferenciación del crecimiento (GDF). La comunicación cruzada entre esta y su contraparte, la vía de TGF-β-activina-nodal, cumple un rol central en numerosos procesos celulares que regulan la proliferación y diferenciación celular. (5)
Las variantes en BMPR2 resultan en una expresión reducida de la proteína funcional, lo que genera la alteración de la transducción de señales del BMP, a menudo junto con un incremento en la respuesta mediada por activina. Actualmente se sabe que este desequilibrio contribuye a la patogenia de la HAP al generar disfunción de las células endoteliales, así como proliferación, resistencia a la apoptosis y contracción de las células musculares lisas vasculares pulmonares. Estos mecanismos resultan en un aumento de las resistencias vasculares pulmonares, incremento de la presión arterial pulmonar y consecuente remodelado del ventrículo derecho. La pérdida del balance entre las vías BMP-GDF y TGF-β-activina-nodal es considerada actualmente el principal defecto molecular con un rol crítico en la predisposición y progresión de la HAP, así como también, un novedoso objetivo terapéutico. (6)
Presentamos el caso de una mujer de 31 años, sin antecedentes médicos, que ingresó a la Sala de Emergencias con disnea progresiva como síntoma principal. La evaluación inicial incluyó una angiotomografía de tórax que descartó tromboembolismo pulmonar agudo y un ecocardiograma Doppler que evidenció hallazgos sugestivos de hipertensión pulmonar (HP): velocidad de regurgitación tricuspídea de 4,02 m/s y disfunción sistólica del ventrículo derecho. La evaluación adicional de la HP incluyó pruebas de función pulmonar, gammagrafía de ventilación-perfusión pulmonar, pruebas de función hepática, serologías virales y colagenograma; todos los resultados fueron dentro de parámetros normales. En la prueba de caminata de 6 minutos la distancia recorrida fue 420 m (59% del valor predicho), con desaturación (del 96% al 86%). En función de dichos resultados se avanzó con un cateterismo cardíaco derecho que demostró una presión media de la arteria pulmonar de 52 mmHg; presión de oclusión de la arteria pulmonar de 6 mmHg; presión media auricular derecha de 2 mmHg; gasto cardíaco de 4,3 L/min; índice cardíaco de 2,95 L/min/m² y resistencia vascular pulmonar de 11 unidades Wood. Se arribó al diagnóstico de HAPI y se inició tratamiento específico con doble terapia: tadalafilo 40 mg/día y ambrisentán 10 mg/día.
Se realizó prueba genética mediante secuenciación del exoma completo como parte de un protocolo en nuestro centro. Se detectó en el gen, una nueva variante heterocigota: NM_001204.7:c.663del (p.Leu222Trpfs*8) localizada en el exón 6. La confirmación se realizó mediante reacción en cadena de la polimerasa (PCR) y secuenciación de Sanger. La variante consiste en una deleción de citosina en la posición 663 que induce un cambio en el marco de lectura y el reemplazo de leucina por triptofano en el codón 222. Esto genera un codón de terminación prematuro 8 tripletes después (Figura 1) que conduce a la síntesis de una proteína BMPR2 más corta, con pérdida de la función. La clasificación de la variante se efectuó según las recomendaciones del American College of Medical Genetics and Genomics. Se determinó que esta variante no fue descripta previamente en la población general ni en pacientes con HAP. En base al análisis realizado se clasificó como variante probablemente patogénica.
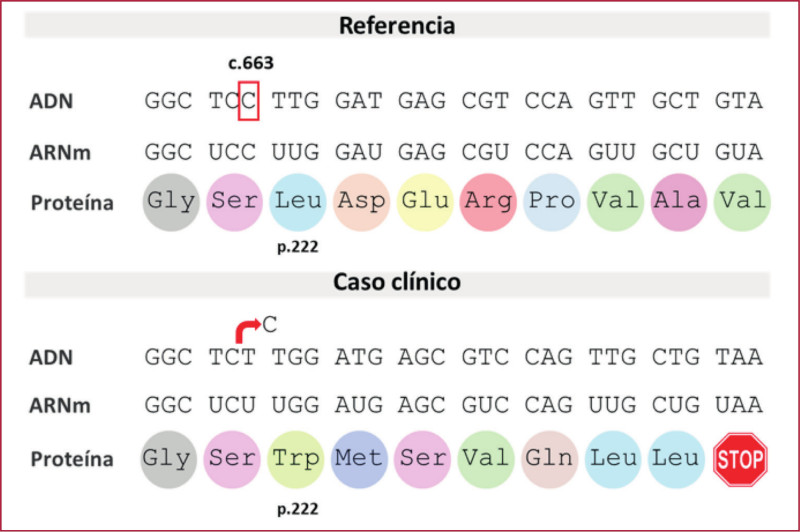
Fig. 1
Análisis de la variante c.663del en el gen BMPR2. Se comparan las secuencias de ADN, ARNm y proteína entre el caso y la secuencia referencia. En el caso se ilustra la deleción de una citosina (C) en el nucleótido 663 (flecha roja) que genera un corrimiento del marco de lectura y forma un codón de terminación prematuro (STOP). Secuencias de referencia BMPR2: NG_009363.1 (gen), NM_001204.7 (transcripto) y NP_001195.2 (proteína).
Con los resultados obtenidos y siguiendo las recomendaciones internacionales actuales se inició la evaluación clínica y genética de los familiares de primer grado. Se efectuó el estudio molecular directo de la variante mediante amplificación del exón 6 por PCR y secuenciación de Sanger en ambos progenitores. El padre de la paciente, quien además al momento del interrogatorio refirió signos y síntomas compatibles, fue diagnosticado con HAP, resultando también positivo en el testeo genético. El diagnóstico de HAP en el padre y la detección de la variante c.663del confirmó la segregación familiar (Figura 2) y avala la reclasificación de esta variante como patogénica. De esta forma, se modificó el diagnóstico de ambos pacientes como portadores de HAPH.
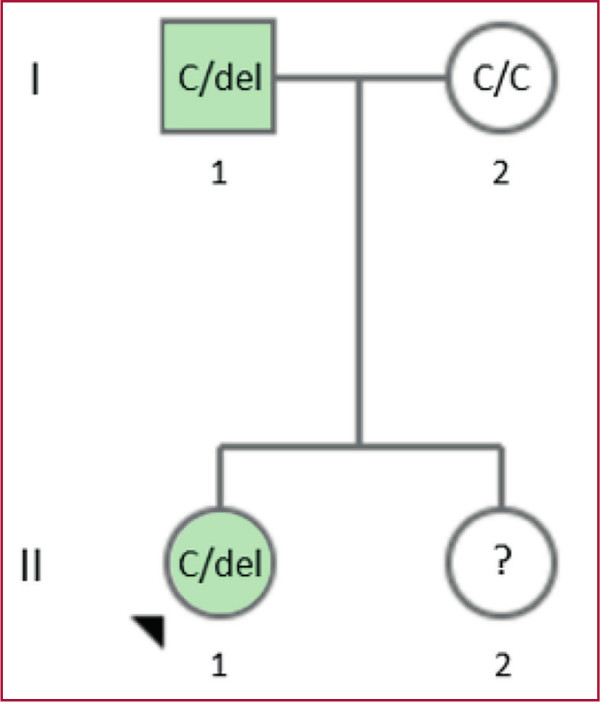
Fig. 2
Árbol genealógico de la paciente con la variante c.663del. Con verde se identifican a los familiares diagnosticados con HAP. La variante se encontró en heterocigosis en el caso índice y su padre (C/del). El genotipo de la madre fue C/C, por lo que no presenta la variante; mientras que la hermana aún no fue estudiada. C: citosina; del: deleción
Los datos obtenidos indican que c.663del es una variante causal de HAP no conocida hasta el momento y constituye además la primera variante de BMPR2 informada en Argentina. La identificación de esta variante permitió confirmar el diagnóstico molecular de enfermedad hereditaria, de importancia tanto para el manejo clínico y terapéutico, así como para el asesoramiento genético de la paciente y sus familiares.
BIBLIOGRAFÍA
Martín de Miguel I, Cruz-Utrilla A, Oliver E, Escribano-Subías P. Novel Molecular Mechanisms Involved in the Medical Treatment of Pulmonary Arterial Hypertension. Int J Mol Sci 2023;24:4147. https://doi.org/10.3390/ijms24044147
Southgate L, Machado RD, Gräf S, et al. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol 2020;17:85-95. https://doi.org/10.1038/s41569-019-0242-x
Cuthbertson I, Morrell NW, Caruso P. BMPR2 mutation and metabolic reprogramming in pulmonary arterial hypertension. Circ Res 2023;132:109-26. https://doi.org/10.1161/CIRCRESAHA.122.321554
Morrell NW, Aldred MA, Chung WK, Elliott CG, et al. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J 2019;53:1801899. https://doi.org/10.1183/13993003.01899-2018.
Guignabert C, Humbert M. Targeting transforming growth factor-β receptors in pulmonary hypertension. Eur Respir J 2021;57:2002341 https://doi.org/10.1183/13993003.02341-2020
Humbert M, Sitbon O, Guignabert C, Savale L, et al. Treatment of pulmonary arterial hypertension: recent progress and a look to the future. Lancet Respir Med 2023;11:804-19. https://doi.org/10.1016/S2213-2600(23)00264-3
Notes
Author notes
Dirección para correspondencia: Josefina Banchio dal Bo. Correo electrónico: banchiojosefina@gmail.com
Conflict of interest declaration
