Comunicaciones
COMPLETE GENOME SEQUENCE OF A RECOMBINANT BRASSICA YELLOWS VIRUS INFECTING Nicotiana tabacum
SECUENCIA COMPLETA DEL GENOMA DE UN VIRUS BRASSICA YELLOWS INFECTANDO A Nicotiana tabacum
SEQUÊNCIA COMPLETA DO GENOMA DE UM VÍRUS BRASSICA YELLOWS INFETANDO A Nicotiana tabacum
COMPLETE GENOME SEQUENCE OF A RECOMBINANT BRASSICA YELLOWS VIRUS INFECTING Nicotiana tabacum
Interciencia, vol. 44, núm. 1, pp. 38-42, 2019
Asociación Interciencia
Recepción: 14 Septiembre 2018
Corregido: 12 Enero 2019
Aprobación: 14 Enero 2019
Financiamiento
Beneficiario: QingFeng Tang, HaoJun Wang and TianSheng
Fuente: Key Program of Anhui Province Tobacco Corporation, China (grant numbers: 20150 551007, 20160551008, 2017055 1024), the Anhui Province Natural Science Foundation, China (grant 1608085QC59), the National Key Research and Development Program of China (grant 2017YFD0200902), the Key Project for Academic and Technical Leader Candidate of Anhui Province, China (Grant 2017H107), and the Innovation Program of Anhui Agricultural University, China (Grant 2018yjs-6)
Abstract: The entire genome of the recombinant brassica yellows virus Anhui (BrYV-AH) was isolated and characterized from tobacco (Nicotiana tabacum) leaves in the Anhui Province, China. Infected leaves were identified by leaf mosaic appearance, yellowing, and deformity. The BrYV-AH sequenced genome is comprised of 5,678 nucleotides (MF314820) and shares 93% nucleotide sequence identity with another brassica yellows virus isolate, the Chinese cabbage-Haidian (KF015269). The sequenced genome contains six open reading frames (ORFs) which encode putative proteins with functions in cell-to-cell movement and suppression of RNA silencing. Phylogenetic analysis places BrYV-AH alongside members of the genus Polerovirus, in the family Luteoviridae. Previous research has reported BrYV infection in tobacco, but the complete genome sequence of the virus had not yet been characterized. To the best of our knowledge, this is the first report of a complete recombinant BrYV genome sequence originating from a tobacco infection.
Keywords: Brassica Yellows Virus, Genome Sequence, Nicotiana tabacum , Recombinant Virus.
Resumen: El genoma completo del virus recombinante brassica yellows Anhui (BrYV-AH) fue aislado y caracterizado a partir de hojas de tabaco (Nicotiana tabacum) en la Provincia de Anhui, China. Se identificaron hojas infectadas por su apariencia de hojas con mosaic, amarillamiento y deformidad. El genoma secuenciado de BrYV-AH está compuesto de 5678 nucleótidos (MF314820) y comparte un 93% de idenbtidad de secuencia con otro aislado de virus brassica yellows, el virus de repollo Haidian chino (KF015269). El genoma contiene seis marcos abiertos de lectura (ORFs) que codifican proteínas putativas con funciones en el movimiento célula-célula y la supresión de silenciamiento por RNA. El análisis filogenético coloca al BrYV-AH junto a miembros del género Polerovirus en la familia Luteoviridae. Investigaciones previas han reportado la infección por BrYV en tabaco, pero el genoma completo no había sido caracterizado. Hasta donde sabemos, este es el primer reporte de la secuencia completa del genoma del BrYV recombinante obtenido de una infección del tabaco.
Resumo: O genoma completo do vírus recombinante brassica yellows Anhui (BrYV-AH) foi isolado e caracterizado a partir de folhas de tabaco (Nicotiana tabacum) na Província de Anhui, China. Identificaram-se folhas infectadas por sua aparência similar a folhas com mosaic, amarelecimento e deformidade. O genoma sequenciado de BrYV-AH está composto de 5678 nucleotídeos (MF314820) e compartilha 93% de identidade de sequência com outro isolado do vírus brassica yellows, o vírus de repolho chinês Haidian (KF015269). O genoma contém seis marcos abertos de leitura (ORFs) que codificam proteínas putativas com funções no movimento célula-célula e a supressão de silenciamento por RNA. A análise filogenética coloca ao BrYV-AH junto a membros do gênero Polerovirus na família Luteoviridae. Investigações prévias têm relatado a infecção por BrYV em tabaco, más o genoma completo não havia sido caracterizado. Até onde sabemos, este é o primeiro relato da sequência completa do genoma do BrYV recombinante obtido de uma infecção do tabaco.
Introduction
Tobacco (Nicotiana tabacum) is an important economic crop worldwide, with half of the world’s tobacco farmers in China, the world’s largest producer (Warner, 2000). The production and yield of tobacco have been seriously affected by the invasion of emerging and recurrent plant viruses with symptoms such as venial necrosis, mosaic, mottling, yellowing, ring spots, stunting, shoestring and deformation (Ding et al., 2010; Wang et al., 2016). Early identification of these plant pathogens remains a focal point in the field of virology, aimed at preventing the spread of the viruses as well as developing ways of combating and reducing their effects on agricultural yield.
Traditional generic methods for identifying and characterizing diseases include the use of enzyme-linked immunosorbent assay (ELISA; Ksiazek et al., 1999), polymerase chain reaction (PCR; Lanciotti et al., 1992), microarray based on prior knowledge of antibody or sequence of the potential virus (Sauder et al., 1996), or techniques such as electron microscopy or use of indicator plants as bioassays (Kreuze et al., 2009). These traditional techniques have now been combined or replaced by molecular or serological assays designed to screen for specific ‘known’ viruses.
Profiling of sRNAs using deep sequencing technologies has helped identify a number of plant viruses that have not been reported previously, and has provided a deeper view of virus populations in plants, which could not be achieved by conventional methods like PCR and ELISA. Deep sequencing is a powerful technology that has been increasingly used for starting the analysis of small interfering RNA (siRNAs) populations, extracted from infected tissues (Giampetruzzi et al., 2012). Viral small silencing RNAs have been found to be produced in plants; they overlap in sequence and can assemble into long contiguous fragments of the invading viral genome from small RNA libraries sequenced by next-generation platforms (Wu et al., 2010a).
Members of the genus Polerovirus are composed of a single, positive-sense genomic RNA covalently linked atthe 5’ to a VPg (viral proteingenome-linked), with no 3’ poly(A) tail (Dreher and Miller, 2006; Zhou et al., 2017). BrYV, like many other poleroviruses, has six open reading frames (ORFs), 5’ and 3’ untranslated regions (UTRs), and an intergenic non-coding region (NCR) between ORF2 and ORF3 (Xiang et al., 2011).
Genetic recombination is critical in the formation of new virus strains (Knierim et al., 2010). Furthermore, it also strongly shapes the genomes of plant RNA viruses (Sztuba-Solińska et al., 2011). ELISA and RT-PCR are typically employed to detect and identify viruses but target only a narrow range of known viruses. Here, we use a combination of deep sequencing of small RNAs and RT-PCR to detect a recombinant Polerovirus infecting N. tabacum.
Material and Methods
Collection and preparation of samples
A field survey of the potential viral pathogens of tobacco was conducted across farm fields in Anhui Province of China. One hundred and three symptomatic (mosaic, mottling, yellowing, ring spots, stunting, shoestring and deformation) leaf samples of cultivated tobacco (Nicotina tobaccum) were collected from different regions of the Anhui province. Leaf samples were immediately frozen in liquid nitrogen and stored at -80ºC untilRNA extraction.
RNA extraction from tobacco leaves
Tobacco leaves exhibiting symptoms of infection were collected from a commercial field in China (Figure 1). Inclusion criteria for symptoms considered: leaf mosaic, yellowing and leaf deformity. Samples were snap frozen in liquid nitrogen and stored at -80C. Samples were pooled at random for total RNA isolation, small RNA (sRNA) library construction, and sequencing. For total RNA extraction, frozen leaves were homogenized in lysis buffer (50mM Tris-HCl, pH 8.0; 150mM LiCl; 5mM EDTA, pH 8.0; 5% SDS). The supernatant was mixed twice with chloroform and RNA was precipitated with isopropanol. Following centrifugation, RNA was re-suspended in nuclease-free water. RNA integrity was verified with an ethidium bromidestained 1.2% agarose gel, and its purity assessed by measuring the absorbance ratio at 260/ 280nm using an Eppendorf Biophotometer plus (Germany). A value of 1.8-2.0 is considered acceptable (Wang et al., 2016).

Figure 1
Symptom of the sample
Recombination analysis
The major sources of variability in RNA viruses are mutations, re-assortments, and recombinations (Worobey and Holmes, 1999). These result in the insertion of unrelated sequence elements, and exchange, duplication, or deletion of existing viral sequence elements (Akinyemi et al., 2016). To detect if genome recombination occurred in BrYV-AH, aligned sequences were examined using the recombination detection program 4 (RDP4; Martin et al., 2015). Nine detection methods from the RDP4 package, including RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, PhylPro, LARD, and 3Seq, were applied.
Bioinformatics analysis
The small RNA library of infected tobacco was constructed and sequenced by the Illumina 2G Analyzer. The nonredundant protein (nr) and nucleotide (nt) sequence databases were downloaded from the NCBI. The Velvet program was downloaded from the European Bioinformatics Institute (EBI). Mapping of small RNAs and assembled contigs to tobacco and viral genomes was done with the BLASTn program using the standard parameters in genome assembly (contigs or viral contig with ≥90% similarity and ≥90% coverage of contigs). Assembled contigs were also examined for similarity of their encoded proteins to databases using the BLASTX program. Additional data analyses were carried out with in-house Perl scripts. The computation analyses were carried out using the campus Genomics Institute Core Facility for Bioinformatics (Wu et al., 2010b).
RT-PCR and Sequencing
The viruses detected by small RNA deep sequencing were characterized further by amplification of partial genome sequences using reverse transcription PCR (RT-PCR). cDNA was synthesized using an Oligo(T)23 primer or random hexamer primers (TaKaRa Biotechnology, Dalian). The RT reaction containing 0.1-02mg of total RNA and Moloney murine leukemia virus(M-MLV) reverse transcriptase (TaKaRa Biotechnology, Dalian), following the manufacturer’s instructions. cDNA was diluted 2-fold and stored at -20C until use. In an effort to generate complete virus genome sequences, specific primers were designed to fill the gaps between siRNA contigs according to the consensus sequences of the specific contigs involved and their relative positions. Rapid amplification of cDNA ends (RACE)-PCR (Takara Biotechnology, Dalian) and Sanger sequencing was performed to obtain the 5’ and 3’ ends of the viral genome. Fragment 1 (position 320 to 1646 nucleotides) was obtained using primers F1: GCTCCCGCCTCC ACCTCCGGTCGTG and R1: GGTAACATCACCTTTGTCG GATTTC. Fragment 2 (position 1308 to 2455 nucleotides) was obtained using primers F2: AACTATAATCTAATGGCA CCAATCC and R2: AGGCGGT AGCGGCCTTCATCGAGCT. Fragment 3 (position 2246 to 4662 nucleotides) was obtained using primers F3: CCACACAC CGTGGGTGGGTAGAAGA and R3: CAGGAAAAATGAT GCATCGGCACCA. The 5’ RACE outer primer used was R03: ACGACCGGAGGTGGAGGC GGGAGCA; and the 3’ RACE outer primer used was F03: AGTCTCCTTTCACGTTGA GACCACT. RT-PCR products were sequenced directly by conventional Sanger sequencing.
Sequence analysis and phylogeny
The open reading frames (ORFs) were identified using the FGENESV0 and ORF finder software, respectively. The nucleotide sequences of BrYV-AH were aligned initially using MUSCLE implemented by MEGA v.5. All sequence alignments were adjusted post hoc by visual inspection to ensure that the alignments were biologically relevant as described in (Morrison, 2006). Phylogenetic reconstructions were obtained by the neighbour-joining method as implemented in the MEGA v.5 program with the above nucleotide substitution models. The support of the internal nodes of the trees was evaluated by the bootstrap method with 1000 replications. Nodes with bootstrap support of <75% were collapsed to the nearest significant node (Wang et al., 2013).
Results and Discussion
Provenance of virus material

Figure 2
Position of contigs on the genome of BrYV.
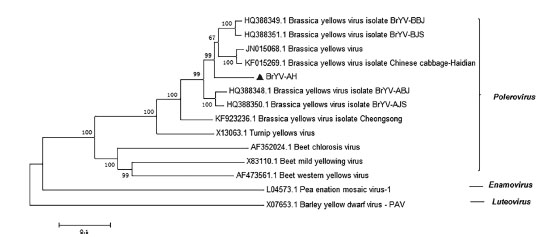
Figure 3
Phylogenetic tree constructed by the neighbor-joining method using MEGA 5, showing the relationship between BrYV-AH and members of the family Luteoviridae. Accession numbers and virus names are given directly in the phylogenetic tree. Values at the nodes show the bootstrap values from 1000 replicates, and the bars represent the evolutionary distances
Sequence properties
The complete genome of BrYV-AH (submitted to GenBank with accession number MF314820) is 5,678 nts in size and the highest nucleotide sequence identity (93%) is shared with the brassica yellows virus isolate known as Chinesecabbage-Haidian (KF015269). BrYV-AH comprises six ORFs, numbered from 0 to 5, which are analogous, both in position and characteristics, to the ORFs in Chinese cabbage-Haidian (Figure 4). No GenBank entry was significantly similar to either the 31 nt long 5’ UTR, or the 186 nt long 3’ UTR of BrYV-AH.
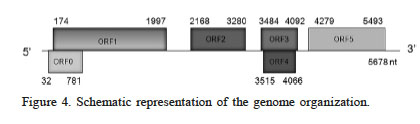
Figure 4
Schematic representation of the genome organization
ORF0 encodes protein P0 (Xiang et al., 2011), which has 100% coverage and 99% identity with P0 of turnip yellows virus (AAL26141.1). P0 is a 28.9kDa protein and contains a putative F-box-like motif that suppresses RNA silencing (Pazhouhandeh et al., 2006). Strong evidence suggests that P0 also determines symptom and host range (Pfeffer et al., 2002). P1 is encoded by ORF1 and shares 100% identity (99% coverage) with P1 (ALL26143.1) of turnip yellows virus. Multiple studies have shown that the P1 protein of potato leaf roll virus plays a critical role in the replication cycle by promoting maturation of the genome-linked virion protein, VPg (Prüfer et al. 1999; Nickel et al. 2008). ORF2 encodesthe putative RNA polymerase (370aa, 41.5kDa, nts 2,168-3,280) and is therefore important for replication. ORF3 (nts 3,484-4,092) encodes a putative 202aa (22.4kDa) major capsid protein (CP) and is followed in-frame by ORF5. The amino acid residues on the surface-oriented loop of the coat protein of Poleroviruses are critical for virus assembly, stability, systemic infection of plants, and movement of virus through aphid vectors (Lee et al., 2005). ORF4 (nts 3,515-4,066) encodes a putative homolog movement protein (183aa, 20.5kDa) with possible functions in cell-to-cell and long-distance movement (Knowles et al., 2012). OFR5 (nts 4,279-5,493) encodes a putative read-through protein (404aa, 45.5kDa). Insect transmission and long distance virus movement through the phloem are carried out by the synergistic work of P3 and P5 (Peter et al., 2009).
Recombination events
Alignment of eleven virus genome sequences (GenBank accession numbers HQ388348, HQ388349, HQ388350, HQ388 351, KF015269, KF923236, KR706247, AF352024, X83110, X13063, and AF473561) with the assembled recombinant strain BrYV-AH (MF314820) revealed significant recombination. The P-values of recombination events recorded byeach of these methods are: RDP (1.178×10-47), GENECONV (1.293×10-28), BootScan (1.701× 10-60), MaxChi (3.360×10-10), Chimaera (4.830×10-23), SiScan (1.533×10-85), and 3Seq (4.954× 10-9). These indicated the presence of recombination events (positions 1-73 and 3677-5928 in the BrYV-AH genome; Figure 5). The potential major parent is AF473561 (beet western yellows virus), and the minor parent is AF352024 (beet chlorosis virus). These results indicate that BrYV-AH is a recombinant virus belonging to the Polerovirus genus.
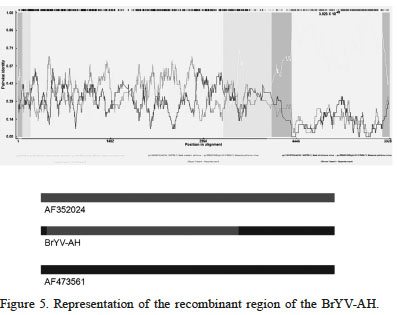
Figure 5
Representation of the recombinant region of the BrYV-AH
Conclusions
In this study we identified a recombinant virus belonging to the genus Polerovirus in the Anhui province of China. We further characterized the genome of BrYV-AH, its variability and the siRNAs induced in tobacco plant in response to virus infection. Our result showed the effectiveness of the custom-made bioinformatics pipeline coupled with molecular techniques and phylogenetic analysis, in diagnostics and identification of plant virus. Survey of plant viruses and prompt diagnostics should be frequently carried out in areas known for large cultivation of economically important crops.
Agradecimientos
his work was supported by the Key Program of Anhui Province Tobacco Corporation, China (grant numbers: 20150 551007, 20160551008, 2017055 1024), the Anhui Province Natural Science Foundation, China (grant 1608085QC59), the National Key Research and Development Program of China (grant 2017YFD0200902), the Key Project for Academic and Technical Leader Candidate of Anhui Province, China (Grant 2017H107), and the Innovation Program of Anhui Agricultural University, China (Grant 2018yjs-6). QingFeng Tang, HaoJun Wang and TianSheng Yang are joint first authors. The authors declare no conflict of interest.
Referencias
Akinyemi IA, Wang F, Zhou B, Qi S, Wu Q (2016) Ecogenomic survey of plant viruses infecting tobacco by Next generation sequencing. Virol. J. 13: 1-12.
Aliyari R, Wu Q, Li HW, Wang XH, Li F, Green LD, Han CS, Li WX, Ding SW (2008) Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in Drosophila. Cell Host Microbe 4: 387-397.
Ding M, Yang C, Zhang L, Jiang ZL, Fang Q, Qin XY, Zhang ZK (2010) Occurrence of Chilli veinal mottle virus in Nicotiana tabacum in Yunnan, China. Plant Dis. 95: 357-357.
Dreher TW, Miller WA (2006) Translational control in positive strand RNA plant viruses. Virology 344: 185-197.
Giampetruzzi A, Roumi V, Roberto R, Malossini U, Yoshikawa N, La Notte P, Terlizzi F, Credi R, Saldarelli P (2012) A new grapevine virus discovered by deep sequencing of virus- and viroid-derived small RNAs in Cv Pinot gris. Virus Res. 163: 262-268.
Knierim D, Deng TC, Tsai WS, Green SK, Kenyon L (2010) Molecular identification of three distinct Polerovirus species and a recombinant Cucurbit aphid-borne yellows virus strain infecting cucurbit crops in Taiwan. Plant Pathol. 59: 991-1002.
Knowles N, Hovi T, Hyypiä T, King A, Lindberg AM, Pallansch M, Palmenberg A, Simmonds P, Skern T, Stanway G (2012) Ninth Report of the International Committee on Taxonomy of Viruses: Picornaviridae . Virus Taxonomy. pp. 105-110.
Kreuze JF, Perez A, Untiveros M, Quispe D, Fuentes S, Barker I, Simon R (2009) Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 388: 1-7.
Ksiazek TG, West CP, Rollin PE, Jahrling PB, Peters CJ (1999) ELISA for the detection of antibodies to Ebola viruses. J. Infect. Dis. 179(Suppl. 1): S192-S198.
Lanciotti RS, Calisher CH, Gubler DJ, Chang GJ, Vorndam AV (1992) Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction. J. Clin. Microbiol. 30: 545-551.
Lee L, Kaplan IB, Ripoll DR, Liang D, Palukaitis P, Gray SM (2005) A surface loop of the potato leafroll virus coat protein is involved in virion assembly, systemic movement, and aphid transmission. J. Virol. 79: 1207-1214.
Letunic I, Doerks T, Bork P (2015) SMART: Recent updates, new developments and status in 2015. Nucl. Ac. Res. 43: D257-D260.
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 1: vev 003-vev003.
Miller JR, Koren S, Sutton G (2010) Assembly algorithms for next-generation sequencing data. Genomics 95: 315-327.
Morrison DA (2006) Multiple sequence alignment for phylogenetic purposes. Aust. Syst. Bot. 19: 479-539.
Nickel H, Kawchuk L, Twyman RM, Zimmermann S, Junghans H, Winter S, Fischer R, Prüfer D (2008) Plantibody-mediated inhibition of the potato leafroll virus P1 protein reduces virus accumulation. Virus Res. 136: 140-145.
Pazhouhandeh M, Dieterle M, Marrocco K, Lechner E, Berry B, Brault V, Hemmer O, Kretsch T, Richards KE, Genschik P, Ziegler-Graff V (2006) F-box-like domain in the polerovirus protein P0 is required for silencing suppressor function. Proc. Natl. Acad. Sci. 103: 1994-1999.
Peter KA, Gildow F, Palukaitis P, Gray SM (2009) The C terminus of the polerovirus P5 readthrough domain limits virus infection to the phloem. J. Virol. 83: 5419-5429.
Pfeffer S, Dunoyer P, Heim F, Richards KE, Jonard G, ZieglerGraff V (2002) P0 of beet Western yellows virus is a suppressor of posttranscriptional gene silencing. J. Virol. 76: 6815-6824.
Prüfer D, Kawchuk L, Monecke M, Nowok S, Fischer R, Rohde W (1999) Immunological analysis of potato leafroll luteovirus (PLRV) P1 expression identifies a 25 kDa RNA-binding protein derived via P1 processing. Nucl. Ac. Res. 27: 421-425.
Sauder C, Müller A, Cubitt B, Mayer J, Steinmetz J, Trabert W, Ziegler B, Wanke K, Mueller-Lantzsch N, de la Torre JC, Grässer FA (1996) Detection of borna disease virus (BDV) antibodies and BDV RNA in psychiatric patients: evidence for high sequence conservation of human blood-derived BDV RNA. J. Virol. 70: 7713-7724.
Sztuba-Solińska J, Urbanowicz A, Figlerowicz M, Bujarski JJ (2011) RNA-RNA recombination in plant virus replication and evolution. Annu. Rev. Phytopathol. 49: 415-443.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28: 2731-2739.
Wang F, Gao ZL, An MN, Zhou BG, Wu YH (2013) Sequencing and phylogenetic analysis of potato virus Y Liaoning isolate in China. J. Integr. Agric. 12: 1195-1200.
Wang F, Qi S, Gao Z, Akinyemi IA, Xu D, Zhou B (2016) Complete genome sequence of tobacco virus 1, a closterovirus from Nicotiana tabacum. Arch. Virol. 161: 1087-1090.
Warner KE (2000) The economics of tobacco: myths and realities. Tobac. Contr. 9: 78-89.
Worobey M, Holmes EC (1999) Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 80: 2535-2543.
Wu Q, Luo Y, Lu R, Lau N, Lai EC, Li WX, Ding SW (2010) Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. 107: 1606-1611.
Xiang HY, Dong SW, Shang QX, Zhou CJ, Li DW, Yu JL, Han CG (2011) Molecular characterization of two genotypes of a new polerovirus infecting brassicas in China. Arch. Virol. 156: 2251-2255.
Yang X, Wang Y, Guo W, Xie Y, Xie Q, Fan L, Zhou X (2011) Characterization of small interfering RNAs derived from the Geminivirus/Betasatellite complex using deep sequencing. PLoS One 6: e16928
Zhou B, Wang F, Zhang X, Zhang L, Lin H (2017) Sequencing and phylogenetic analysis of tobacco virus 2, a polerovirus from Nicotiana tabacum. Arch. Virol. 162: 2159-2162.
Notas de autor
913678797@qq.com