Notas clínicas
DOI: https://doi.org/10.60147/b899ce54
Resumen: El síndrome de Stickler es una enfermedad genética rara del tejido conjuntivo, con herencia autosómica dominante la mayoría de las veces. Se caracteriza por la asociación de oftalmopatía (miopía, desprendimiento de retina y cataratas), anomalías orofaciales, hipoacusia y artropatía, con una amplia variabilidad en la expresión clínica. La confirmación diagnóstica se obtiene por estudio genético. El pronóstico varía en función de la gravedad de las manifestaciones. La ausencia de un tratamiento etiológico reduce las posibilidades terapéuticas a un tratamiento sintomático que muy frecuentemente es quirúrgico. La importancia de conocer sus características radica en una sospecha clínica precoz para tratar de evitar secuelas irreversibles en edades tempranas, y brindar a la familia un asesoramiento genético adecuado.
Palabras clave: Catarata, Colagenosis hereditaria, Desprendimiento de retina, Síndrome de Stickler.
Abstract: Stickler's syndrome is a rare genetic disease of connective tissue, with dominant autosomal inheritance most of the time. It is characterized by the association of ophthalmopathy (myopia, retinal detachment and cataracts), orofacial abnormalities, hearing loss and arthropathy, with a wide variability in clinical expression. Diagnostic confirmation is obtained by genetic study. The prognosis varies depending on the severity of the manifestations. The absence of an etiological treatment reduces the therapeutic possibilities to a symptomatic treatment that is very often surgical. The importance of knowing its characteristics lies in an early clinical suspicion to try to avoid irreversible sequelae at an early age and provide the family with adequate genetic counseling.
Key words: Cataract, Hereditary collagenosis, Retinal detachment, Stickler syndrome.
CASO CLÍNICO
Niño de 5 años que fue traído a consulta tras objetivar, el día previo en domicilio, una mancha blanca en el ojo izquierdo. A la exploración se apreció leucocoria en ojo izquierdo y, tras la medición de la agudeza visual (AV) con optotipos presentó AV de 0,5 en ojo derecho y de 0 en ojo izquierdo, refería ver todo negro. Se realizó valoración oftalmológica urgente que confirmó la presencia de catarata total y desprendimiento de retina izquierda. Dado el daño visual irreversible y la aparición posterior de dolor ocular, se realizó evisceración del globo ocular izquierdo y colocación de una prótesis ocular. Fenotípicamente, impresionó de leve retrognatia, sin otras anomalías craneofaciales ni esqueléticas. Antecedente de miringotomía y colocación de drenajes transtimpánicos a los 8 meses por otitis media aguda recurrentes. Portador de audífonos por hipoacusia bilateral. Sin antecedentes familiares de oftalmopatía ni patología auditiva conocidos.
Dados los hallazgos clínicos, se consideró la posibilidad de que pudiera tratarse de una colagenosis (enfermedad de Stickler o Wagner) y se realizó estudio genético, en el que fueron secuenciados los genes COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, COL9A3 y VCAN, encontrándose la mutación c.1597C>T en el gen COL2A1, que produce un codón de parada (p.R533X; proteína truncada), descrita como patológica, lo que permitió confirmar el diagnóstico de síndrome de Stickler. Se propuso estudio genético familiar.
DISCUSIÓN
El síndrome de Stickler es una enfermedad genética rara del tejido conjuntivo de herencia autosómica, mayoritariamente dominante1,2. Pertenece al grupo de las colagenosis hereditarias2. Fue descrito como una artroftalmopatía progresiva hereditaria, en 1965, por Gunarr B. Stickler2. Se estima que la incidencia al nacimiento es de 1/7500 - 1/90 001. Es la causa hereditaria más frecuente de desprendimiento de retina en la infancia3,4.
Existen 40 genes diferentes que codifican como mínimo a 27 tipos de colágeno. El síndrome de Stickler se debe a una mutación muy heterogénea en los genes que controlan la síntesis del colágeno 2, 9 y 11, por lo que la expresión fenotípica es muy variable2. La transmisión del síndrome en general sigue un modo de herencia autosómico dominante, excepto las mutaciones en los genes del colágeno 9, que siguen un patrón autosómico recesivo1.
Se clasifica en varios tipos según el gen mutado. El tipo I, producido por la mutación en el gen COL2A1, como ocurre en el caso presentado, es el más frecuente2. En él predomina la patología oftálmica, con un mínimo o ninguna característica sistémica3.
El síndrome de Stickler se caracteriza por una combinación variable de manifestaciones1: oculares (cataratas precoces, miopía alta, desprendimiento de retina, glaucoma); craneofaciales, las cuales son de gravedad variable y no están sistemáticamente presentes (úvula bífida, paladar hendido aislado o formando parte de una secuencia de Pierre Robin, hipoplasia malar, microrretrognatia); osteomioarticulares (displasia espondiloepifisiaria, escoliosis, cifosis, espondilolistesis, epifisiólisis de la cabeza femoral, osteonecrosis avascular de la cabeza femoral, artrosis precoz secundaria a una hiperlaxitud… entre otras); y otológicas (hipoacusia de transmisión por disfunción de la trompa de Eustaquio o anomalías en la cadena de huesecillos, hipoacusia neurosensorial, hipoacusia mixta y otitis media). Puede asociar prolapso de la válvula mitral. Se sugiere que debe tenerse en cuenta este diagnóstico si existen hallazgos clínicos en dos o más de las categorías descritas2. Se recomienda el estudio genético tras confirmar clínicamente la sospecha.
En el año 2005, se propusieron los criterios diagnósticos para el síndrome de Stickler tipo I5, descritos en la Tabla 1.
Tabla 1. Criterios diagnósticos del síndrome de Stickler tipo I
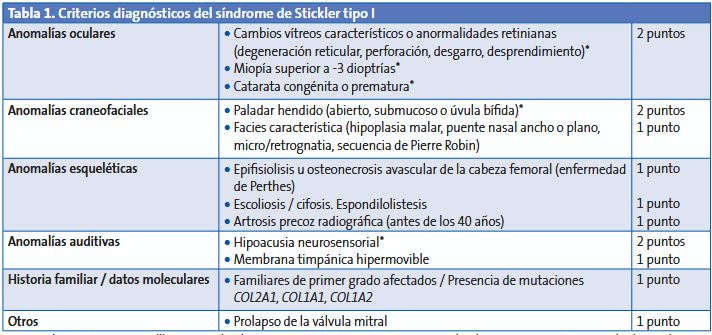
Debe hacerse diagnóstico diferencial con otras colagenosis hereditarias que asocien alteraciones visuales severas en la infancia o desprendimiento de retina precoz (por ejemplo, síndrome de Wagner)2,3.
Ante la ausencia de un tratamiento etiológico, el único tratamiento disponible es el sintomático, que exige una actuación multidisciplinar. El pronóstico varía en función de la gravedad de las manifestaciones1. La importancia del conocimiento de este síndrome para un diagnóstico precoz radica en la prevención de secuelas irreversibles (el desprendimiento de retina es causa de ceguera en hasta el 4% de los casos)3, recomendándose controles anuales que permitan detectar prematuramente las posibles complicaciones2 e implementar el tratamiento correspondiente. El objetivo es aumentar la probabilidad de una evolución más favorable3.
BIBLIOGRAFÍA
El síndrome de Stickler. Artro-oftalmopatía hereditaria progresiva. En: Enciclopedia Orphanet de la Discapacidad [en línea] [consultado el 25/02/2024]. Disponible en www.orpha.net/data/patho/Han/Int/es/SindromeStickler_Es_es_HAN_ORPHA828.pdf
Riera Matute G, Riera Alonso E. Síndrome de Stickler. Semin Fund Esp Reumatol. 2009;10(3):83-6. https://doi.org/10.1016/S1577-3566(09)72129-3
Oscullo Yepez VR, Sierra Santos l, Oscullo Yepez JJ, Oscullo Yepez GJ. Síndrome de Stickler: cuando el desprendimiento de retina se hereda. Rev Clin Med Fam. 2018;11(2):112-5.
Vilaplana F, Muiños SJ, Nadal J, Elizalde J, Mojal S. Síndrome de Stickler. Epidemiología del desprendimiento de la retina. Arch Soc Esp Oftalmol. 2015;90(6):264-8. https://doi.org/10.1016/j.oftal.2014.11.001
Rose PS, Levy HP, Liberfarb RM, Davis J, Szymko-Bennett Y, Rubin B, et al. Stickler syndrome: clinical characteristics and diagnostic criteria. Am J Med Genet A. 2005;138A(3):199-207. https://doi.org/10.1002/ajmg.a.30955
Glossary
AV: agudeza visual.
Referencia para citar este artículo:
CONFLICTO DE INTERESES
RESPONSABILIDAD DE LOS AUTORES
Los autores han remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
