Reporte de casos
A c.3037G>A mutation in FBN1 gene causing Marfan syndrome with an atypically severe phenotype
Mutación c.3037G>A en el gen FBN1 causa sindrome de Marfan con fenotipo atípico severo
A c.3037G>A mutation in FBN1 gene causing Marfan syndrome with an atypically severe phenotype
Investigación Clínica, vol. 58, no. 1, pp. 70-78, 2017
Universidad del Zulia
Received: 29 November 2015
Accepted: 30 November 2016
Abstract: Marfan syndrome is a pleiotropic connective tissue disease inherited as an autosomal dominant trait, mostly caused by mutations in the FBN1 gene, which is located on chromosome 15q21.1 and encoding fibrillin 1. We report a case of Marfan syndrome presenting with severe ocular and systemic manifestations, such as cardiac congenital anomalies. The patient underwent a multidisciplinary approach and his clinical diagnosis was associated with a c.3037G>A mutation in the FBN1 gene. Identification of this genetic alteration should instigate a prompt multidisciplinary assessment and monitoring, in order to prevent devastating consequences such as cardiac and ocular phenotype. Molecular modeling of the mutation highlighted the importance of the preservation of the calcium-dependent structure of an epidermal-growth-factor-like domain of fibrillin-1 and consequently the microfibrillar formation process. This report aims to highlight the importance of an early clinical and molecular diagnosis and once more, the importance of the multidisciplinary approach of this genetic entity.
Keywords: Marfan syndrome, c.3037G>A, FBN1, phenotype.
Resumen: El síndrome de Marfan es una enfermedad pleitrópica del tejido conjuntivo que exhibe un patrón de herencia autosómico dominante, en su mayoría causado por mutaciones en el gen FBN1, que se encuentra en el cromosoma 15q21.1 y codifica a la fibrilina 1. Se presenta un caso de síndrome de Marfan que cursa con manifestación sistémica severa cardíaca y principlamente ocular. El paciente presentó una valoración multidisciplinaria y su diagnóstico clínico fue asociado con la mutación c.3037G>A en el gen FBN1. La identificación de esta alteración genética debe promover una pronta evaluación y supervisión con el fin de evitar las desvastadoras consecuencias, tales como el fenotipo cardíaco y ocular. El modelado comparativo de proteínas resalta la importancia de la conservación de la estructura del dominio de la fibrilina-1 dependiente de calcio similar al factor de crecimiento epidérmico y por lo tanto el proceso de formación microfibrilar. Este informe tiene como objetivo resaltar la importancia de un diagnóstico clínico y molecular temprano y el enfoque multidisciplinario de esta entidad genética.
Palabras clave: síndrome de Marfan, c.3037G>A, FBN1, manifestaciones oculares.
INTRODUCTION
Marfan syndrome (MFS, OMIM #154700) is a rare genetic pleiotropic disorder, presenting with skeletal, ocular, skin, and cardiovascular symptoms. First described by the French pediatrician Antoine Bernard-Jean Marfan in 1896, is an inherited connective tissue disease mostly transmitted as an autosomal dominant trait resulting from mutations in the FBN1gene (OMIM #134797). This gene contains 66 exons, is located on chromosome 15q21.1 and encodes fibrillin, an important protein of the extracellular matrix, that contributes to the final structure of a microfibril (1, 2). Estimated incidences range between 1:5,000 to 10,000 live births and no gender or ethnic associations have been reported (2).
Three international nosologies have been proposed for the diagnosis of MFS, the Berlin nosology in 1988, was purely based on the clinical phenotype. The Ghent nosology in 1996 (Ghent-1), which was a revision of the Berlin criteria, used the discovered FBN1 mutations as a component in the diagnostic criteria, and subsequently, the revised Ghent nosology in 2010 (Ghent-2), highlighted the FBN1 mutation, aortic dilatation and “ectopia lentis” in the diagnosis of this entity (3, 4, 5). Moreover, mutations in the FBN1 gen can result in classic MFS, neonatal MFS, ectopia lentis, familial ascending aortic aneurysm and isolated skeletal abnormalities. These conditions are collectively termed type-1 fibrillinopathies. There is a remarkable degree of clinical variability both intra- and inter-family with FBN1 mutations, which has made the investigation of the genotype–phenotype correlations difficult (6). However, molecular genetic testing of FBN1 is central to diagnosis, genetic counselling and to document a potential genotype-phenotype correlation (7). We report case of a male patient with a heterozygous missense mutation c.3037G>A in the FBN1 gene with MFS presenting severe cardiac and mainly ocular phenotype.
CLINICAL REPORT
Here we report the case of a 25-year-old Italian male with MFS, presenting severe cardiac and mainly ocular phenotype. From available medical records: at birth arthrogryposis and excessive length were seen, associated with transient oxygen requirement due to poor respiratory adaptation of the newborn, that improved in the first two weeks of life. During the first two years of life, high stature, and mild motor retardation, characterized by difficult walking were suggestive of MFS. Subsequently, several anomalies were evidenced: narrow palate, pectus excavatum, recurrent hernias, arachnodactyly, general marfanoid physical aspect, and joint laxity with a high degree of elbow extension, besides cardiac and ocular anomalies.
Echocardiography revealed mild dilatation of ascending aorta, associated with mild aortic regurgitation in the first year of age. Though asymptomatic, the diameter of the ascending aorta progressed up to 4 cm with clear aneurysm evolution in few years; for this reason, the patient underwent elective ascending aorta replacement and conservative aortic valve surgery at age 5. During subsequent years, aortic valve regurgitation recurred and worsened, together with mitral valve prolapse and regurgitation. The patient was treated with beta-blockers and ace-inhibitors for some years, waiting for complete pubertal development. When he was 16 years old the aortic and mitral dysfunction had become severe, with volume remodeling of the left ventricle, left atrium enlargement and severe bivalvular regurgitations. After discussion about the surgical options, the patient chose valve replacement with mechanical prosthesis, and chronic oral anticoagulants. The surgeon was able to repair the mitral valve and substituted the aortic valve with a mechanical prosthesis.
Concerning the ocular history the patient presented bilateral posterior lens dislocation in the first year of life. In the first decade the patient underwent lensectomy, vitrectomy and scleral buckling in each eye for total retinal detachment. His axial lengths were 33.0 mm right and 32.5 mm left. Silicone oil tamponade was required due to recurrent retinal detachment. The patient subsequently developed in silicone oil keratopathy with corneal endothelial failure and decompensation in the right eye requiring two penetrating keratoplasties, at ages 21 and 23, respectively. His visual acuities were no perception of light in the right eye and hand movements in the left eye. The refraction was +3.50 +1.00 at 70° in the right eye and +4.00 +1.00 at 10° in the left eye. Slit-lamp examination was performed at age 25 and demonstrated bilateral aphakia, corneal opacity with peripheral neovascularization in the right eye and a transparent cornea in the left eye. The intra-ocular pressure was 14 mmHg in each eye as measured by Goldmann applanation tonometry. Fundus examination showed bilateral persistent retinal detachment in each eye (Fig. 1 a,b).
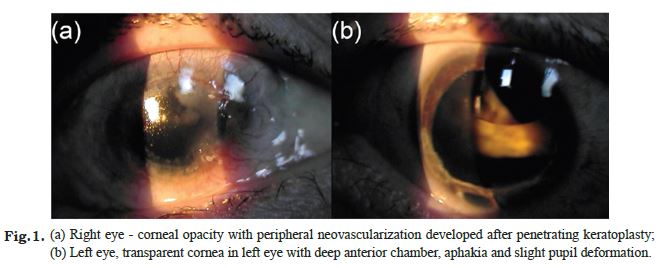
Fig. 1 a,b
Fig. 1 a,b
Fig.1. (a) Right eye - corneal opacity with peripheral neovascularization developed after penetrating keratoplasty; (b) Left eye, transparent cornea in left eye with deep anterior chamber, aphakia and slight pupil deformation.
Clinical and radiological dental examination at 20 years age revealed dental defects such as macrodontia and he had been treated with current recommendations. The patient was not affected by mental retardation, he completed university studies until graduation, spite of his total blindness status.
The DNA extracted from peripheral blood and Sanger sequencing, demonstrated a heterozygous missense mutation c.3037G>A in exon 24 of the FBN1 gene (NM_000138) resulting in a non-conservative substitution of a glycine residue for arginine (p.G1013R). Homology modeling of fibrillin-1 in the 909-1069 amino acid region was made with the program MODELLER (9v10) (8), using as the template the structure of the calcium-bound fragment (residues 1486-1647) of the same protein (Protein Data Bank, PDB, entry 1UZK), according to the sequence alignment shown in (Fig. 2).
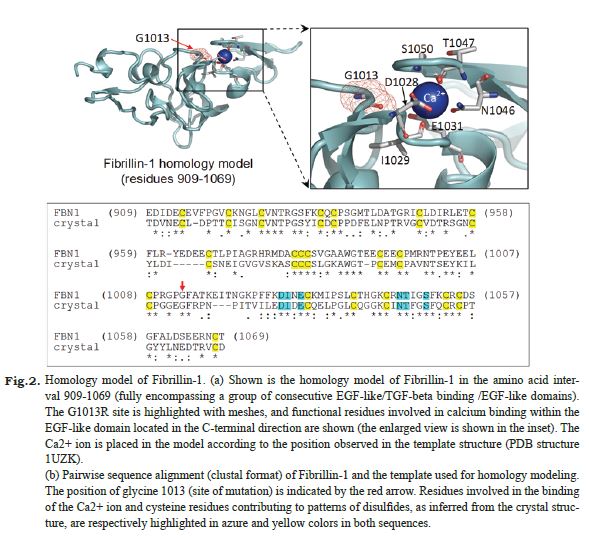
Fig. 2.
Fig. 2. Homology model of Fibrillin-1.
(a) Shown is the homology model of Fibrillin-1 in the amino acid interval 909-1069 (fully encompassing a group of consecutive EGF-like/TGF-beta binding /EGF-like domains). The G1013R site is highlighted with meshes, and functional residues involved in calcium binding within the EGF-like domain located in the C-terminal direction are shown (the enlarged view is shown in the inset). The Ca2+ ion is placed in the model according to the position observed in the template structure (PDB structure 1UZK).
(b) Pairwise sequence alignment (clustal format) of Fibrillin-1 and the template used for homology modeling. The position of glycine 1013 (site of mutation) is indicated by the red arrow. Residues involved in the binding of the Ca2+ ion and cysteine residues contributing to patterns of disulfides, as inferred from the crystal structure, are respectively highlighted in azure and yellow colors in both sequences.
DISCUSSION
The patient studied here had an atypically severe MFS, according Tiecke et al. (6), in which the clinical peculiarities are the extraordinary early manifestations of severe aortic anomalies, leading to aneurysm development in the first years of life, together with the relatively early, but severely evolving ophthalmologic anomalies, in a sporadic case. Based on its clinical course and severity, differential diagnoses with Loyes Dietz syndrome could be discussed, but genetic molecular findings definitely excluded, confirming the diagnosis of MFS associated with (c.3037G>A; p.G1013R) mutation in the FBN1gene. This mutation was previously reported in three unrelated patients with severe and atypically severe manifestations, with neonatal presentation (6, 7). The mutation affects a highly conserved residue in an interdomain linkage region and was first reported by Nijbroek et al (7), in a patient with neonatal presentation. Tiecke et al. (6), identified this mutation by heteroduplex screening in two unrelated patients with severe clinical involvement, who were not members of their initial screening group. In Table I, is described a comparative clinical feature between the patients studied with the same mutation.
| Findings/Authors | Nijbroek et al7 | Tiecke et al 6 | Present case | |
| Sex | F | F | F | M |
| Inheritance | Sporadic | Sporadic | Sporadic | Sporadic |
| Organ/System | ||||
| Skeletal | Severe deformity Marked joint contractures | Kyphoscoliosis Pectus excavatum Arachnodactyly | na | Pectus excavatum Arachnodactyly |
| Thin (weight < 3th percentile and height > 97th percentile) | Tall (height > 97th percentile) | |||
| Ocular | Ectopia lentis | Complete luxation of both lentis | Bilateral ectopia lentis | Bilateral posterior ectopia lentis |
| Progressive and severe myopia | Bilateral aphakia | |||
| Corneal endothelial failure | ||||
| Bilateral retinal detachment | ||||
| Cardiovascular | Severe aortic dilatation | Dilatation of the aortic root | ||
| Panvalvular dysfunction | Second-degree mitral insufficiency | Dilatation of ascending aorta | Dilatation of ascending aorta with aneurysm | |
| Congestive heart failure | Panvalvular dysfunction | Panvalvular dysfunction | ||
| Integument Others | Crumpled ears | Parchment-like skin | na | Mild aortic regurgitation |
| Recurrent hernia | ||||
| Narrow palate | ||||
| Macrodontia | ||||
| Form | Atypically severe | Severe | Severe | Atypically severe |
| Neonatal presentation | Diagnosed at the age of 8 months | Arthrogryposis neonatal | ||
| Evolution | Functionally crippled | Died due to suture dehiscence and internal hemorrhage | na | Motor retardation |
| Circulatory insufficiency | No Mental retardation | |||
| Devastating joint laxity and contracture | ||||
Among cardiovascular manifestations, mitral valve dysfunction, dilation of the ascending aorta and aneurysm are the most frequent. Aortic dilation is present in 35% of patients prior to 5 years of age and up to 70% of patients prior to 20 years of age. Aortic pathology often necessitates surgical measures, including aortic root replacement at an early age (9, 10). These conditions were present at an early age in the patient studied.
The main ocular manifestations in MFS are myopia and “ectopia lentis”. The subluxation or luxation of the lens is reported in approximately 45 – 87% (11) of the cases. Börger found “ectopia lentis” to be part of MFS in 1914, and this entity is reported to be its most prevalent cause. In a ocular study in 87 patients with MFS, the “ectopia lentis” was found in 108 eyes (62.1%). Moreover, of the 68 phakic eyes with “ectopia lentis”, 43 (63.2%) had subluxation. Myopia above 3D occurred in 38.4% of the phakic eyes (12). Other ocular findings includes glaucoma secondary to aphakia, abnormally flat cornea, increased axial length of the globe, hypoplastic iris, orciliary muscle responsible for decreased miosis, and an increased risk of retinal detachment (11).
Retinal detachment is a serious complication of MFS occurring in 5-11% of patients (13). The frequency increases to 8 – 38% in the presence of “ectopia lentis” (13, 14). Moreover, myopia, previous intraocular surgery, and increased axial length are associated with a high risk of retinal detachment (15). MFS patients are also more prone to develop retinal detachment because of unstable subluxated or dislocated lens which exerts traction on the vitreous base leading to small tears or holes in the periphery of the retina. Scleral buckling is recommended as a first surgical procedure (13), if the crystalline lens is normally placed and vitrectomy and internal tamponade is required in the presence of failed scleral buckling, proliferative vitreoretinopathy, a posteriorly dislocated lens, a subluxated or cataracts lens, not allowing an adequate evaluation of the fundus periphery and giant retinal tears (16,17). In the case mentioned here, bilateral spontaneous complete posterior lens dislocation developed in early childhood, bilateral total rhegmatogenous retinal detachment persisted in spite of repeated vitrectomies and internal tamponade. Corneal decompensation followed silicone oil keratopathy and two penetrating keratoplasties were performed. Prompt and aggressive treatment of ocular complication failed to prevent more severe visual loss and was unable to improve the quality of the patient’s life.
The FBN1gene encodes for fibrillin-1, which is a 350 kDa glycoprotein member of the fibrillin family, that are the major components of microfibrils in the extracellular matrix of elastic and non-elastic tissues. These proteins have a modular structure composed of several epidermal-growth-factor-like (EGF) domains (both the type with calcium-binding ability and the type unable to bind calcium are present) and a number of transforming growth factor (TGF)-beta binding domains, which contribute to the structure of 10-12 nm thick microfibrils in the extracellular matrix in a calcium dependent manner (18). The (c.3037G>A; p.G1013R) mutation falls in a peptide linker between a TGF-beta binding domain and an EGF-like domain, and to investigate the possible effects of this mutation on the protein we built the structural model of the 909-1069 amino acid region of fibrillin-1 (encompassing a group of three consecutive EGF-like/TGF-beta binding/EGF-like domains), based on the homology (44% amino acid identity) shared with the structurally characterized amino acid region 1486-1647 of the same protein. These two regions of fibrillin-1 also share same domain architecture, and this allowed modeling the inter-domain linker hosting the site of the p.G1013R mutation in the context of its interactions with nearby domains. In fact, as shown in (Fig. 2), the glycine 1013-containing linker is structured and packed against the calcium-bound structure of the EGF-like domain adjacent on the C-terminal side, and in particular glycine 1013 contributes to a turn and is proximal to the calcium-binding residues of this domain. The replacement of the neutral and tiny glycine 1013 residue with the much bulkier and positively charged arginine in the p.G1013R is expected to produce both structural and electrostatic perturbations, which are expected to impair the calcium-depending structure of an EGF-like domain of fibrillin-1 and likely to interfere with the process of microfibril formation.
In conclusion, the c.3037G>A mutation in FBN1 has been previously reported as pathogenic, resulting in either severe or atypically severe or neonatal forms of MFS (6, 7). The case reported herein highlights severe cardiac problems encountered and the devastating ocular phenotype associated with this specific FBN1 mutation. Identification of this mutation should instigate a prompt cardiac and ophthalmological assessment and monitoring, in order to intervene with ophthalmic care prior to severe ophthalmic consequences. Molecular modelling identified that the glycine residue at position 1013 is crucial to preserve the calcium-dependent structure of an EFG-like domain of fibrillin-1 and consequently the microfibrillar formation process.
References
1. Pepe G, Giusti B, Sticchi E, Abbate R, Gensini GF, Nistri S. Marfan syndrome: current perspectives. Appl Clin Genet 2016; 9:55-65.
2. Verstraeten A, Alaerts M, Van Laer L, Loeys B. Marfan syndrome and related disorders: 25 Years of Gene Discovery.Hum Mutat 2016;37:524-531.
3. von Kodolitsch Y, De Backer J, Schüler H, Bannas P, Behzadi C, Bernhardt AM, Hillebrand M, Fuisting B, Sheikhzadeh S, Rybczynski M, Kölbel T, Püschel K, Blankenberg S, Robinson PN. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl Clin Genet 2015;8:137-155.
4. Groth KA, Hove H, Kyhl K, Folkestad L, Gaustadnes M, Vejlstrup N, Stochholm K, Østergaard JR, Andersen NH, Gravholt CH. Prevalence, incidence, and age at diagnosis in Marfan syndrome. Orphanet J Rare Dis 2015; 10:153.
5. Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47:476-485.
6. Tiecke F, Katzke S, Booms P, Robinson PN, Neumann L, Godfrey M, Mathews KR, Scheuner M, Hinkel GK, Brenner RE, Hövels-Gürich HH, Hagemeier C, Fuchs J, Skovby F, Rosenberg T. Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype- phenotype correlations in FBN1 exons 24-40. Eur J Hum Genet 2001; 9:13-21.
7. Nijbroek G, Sood S, McIntosh I, Francomano CA, Bull E, Pereira L, Ramirez F, Pyeritz RE, Dietz HC. Fifteen novel FBN1 mutations causing Marfan syndrome detected by heteroduplex analysis of genomic amplicons. Am J Hum Genet 1995; 57:8-21.
8. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993; 234:779-815.
9. Ware AL, Miller DV, Erickson LK, Menon SC. Marfan syndrome associated aortic disease in neonates and children: a clinical-morphologic review. Cardiovasc Pathol 2016; 25:418-422.
10. Treasure T, Takkenberg JJ, Pepper J. Surgical management of aortic root disease in Marfan syndrome and other congenital disorders associated with aortic root aneurysms. Postgrad Med J 2016; 92(1084):112-117.
11. Latasiewicz M, Fontecilla C, Millá E, Sánchez A. Marfan syndrome: ocular findings and novel mutations-in pursuit of genotype-phenotype associations. Can J Ophthalmol 2016;51:113-118.
12. Drolsum L, Rand-Hendriksen S, Paus B, Geiran OR, Semb SO. Ocular findings in 87 adults with Ghent-1 verified Marfan syndrome. Acta Ophthalmol 2015;93:46-53.
13. Nahum Y, Spierer A. Ocular features of Marfan syndrome: diagnosis and management. Isr Med Assoc J 2008; 10:179-181.
14. Dean JC. Marfan syndrome: clinical diagnosisand management. Eur J Hum Genet 2007; 15:724-733.
15. Wheatley HM, Traboulsi EI, Flowers BE, Maumenee IH, Azar D, Pyeritz RE, Whittum-Hudson JA. Immunohistochemical localization of fibrillin in human ocular tissues. Relevance to the Marfan syndrome. Arch Ophthalmol 1995; 113:103-109.
16. Sharma T, Gopal L, Shanmugam MP, Bhende PS, Agrawal R, Shetty NS, Gopalakrishna M, Rao MK, Balusamy S. Retinal detachment in Marfan syndrome: clinical characteristics and surgical outcome. Retina 2002; 22:423-428.
17. Remulla JF, Tolentino FI. Retinal detachment in Marfan’s syndrome. Int Ophthalmol Clin 2001; 41:235-240.
18. Zeyer KA, Reinhardt DP. Engineered mutations in fibrillin-1 leading to Marfan syndrome act at the protein, cellular and organismal levels. Mutat Res Rev Mutat Res 2015; 765:7-18.
Author notes
mcallea@gmail.com