Casos clínicos
DEFICIENCIA DE LA ENZIMA 17ALFA-HIDROXILASA CON HIPERALDOSTERONISMO
17ALPHA-HYDROXYLASE DEFICIENCY WITH HYPERALDOSTERONISM
DEFICIENCIA DE LA ENZIMA 17ALFA-HIDROXILASA CON HIPERALDOSTERONISMO
Revista Venezolana de Endocrinología y Metabolismo, vol. 20, núm. 2, pp. 107-112, 2022
Sociedad Venezolana de Endocrinología y Metabolismo

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 3.0 Internacional.
Recepción: 01 Junio 2021
Aprobación: 01 Febrero 2022
Resumen:
Objetivo: Presentar un caso de deficiencia de 17alfa-hidroxilasa con hiperaldosteronismo.
Caso clínico: Se trata de paciente femenina de 16 años de edad quien presentó amenorrea primaria acompañada de ausencia de caracteres sexuales secundarios. Al examen físico se encontraron cifras tensionales elevadas y ausencia de vello púbico y axilar. Las pruebas de laboratorio mostraron valores de potasio sérico bajo con sodio normal. Las pruebas hormonales demostraron aumento de las concentraciones de hormona adenocorticotropa, 11-deoxicorticosterona y gonadotropinas, con disminución de cortisol, sulfato de dehidroepiandrosterona, 17alfa-hidroxiprogesterona y esteroides sexuales. También se encontraron valores de aldosterona elevados con baja actividad de la renina plasmática. Los estudios por imágenes demostraron útero rudimentario, ovarios pequeños y glándulas suprarrenales normales. El análisis genético encontró cariotipo normal (46 XX) con mutación patogénica en el exón 6 del CYP17A1 confirmando el diagnóstico de deficiencia de 17alfa-hidroxilasa. Las manifestaciones clínicas desaparecieron luego del tratamiento con glucocorticoides y hormonas sexuales.
Conclusión: La deficiencia de 17alfa-hidroxilasa es una forma rara de hiperplasia suprarrenal congénita con un patrón de herencia autosómico recesivo, en la que los defectos en la biosíntesis del cortisol y los esteroides sexuales dan lugar a un exceso de mineralocorticoides. Los casos que presentan hiperaldosteronismo son muy raros y están probablemente asociados con la severidad de la deficiencia enzimática. Esta condición debe sospecharse en sujetos jóvenes con hipertensión primaria, hipokalemia y ausencia de los caracteres sexuales secundarios. La identificación de la mutación puede ayudar a una mejor comprensión de la enfermedad.
Palabras clave: Deficiencia de 17alfa-hidroxilasa, hiperaldosteronismo, hiperplasia suprarrenal congénita, hipertensión, hipokalemia.
Abstract:
Objective: To present a case of 17alpha-hydroxylase deficiency with hyperaldosteronism.
Clinical case: A 16-year-old female patient presented with primary amenorrhea accompanied by absence of secondary sexual characteristics. Physical examination revealed elevated blood pressure and absence of pubic and axillary hair. Laboratory tests showed low serum potassium values with normal sodium. Hormonal tests showed increased concentrations of adrenocorticotropic hormone, 11-deoxycorticosterone and gonadotropins with decreased cortisol, dehydroepiandrosterone sulfate, 17alpha-hydroxyprogesterone and sex steroids concentrations. Aldosterone values were also elevated with low plasma renin activity. Imaging studies demonstrated rudimentary uterus, small ovaries and normal adrenal glands. Genetic analysis found normal karyotype (46 XX) with pathogenic mutation in exon 6 of CYP17A1 confirming the diagnosis of 17alpha-hydroxylase deficiency. Clinical manifestations disappeared after treatment with glucocorticoids and sex hormones.
Conclusion: 17alpha-hydroxylase deficiency is a rare form of congenital adrenal hyperplasia with an autosomal recessive inheritance pattern, in which defects in cortisol and sex steroid biosynthesis result in mineralocorticoid excess. Cases presenting with hyperaldosteronism are very rare and are probably associated with severity of the enzyme deficiency. This condition should be suspected in young subjects with primary hypertension, hypokalemia and absence of secondary sexual characteristics. Identification of the mutation may help to better understand the disease.
Keywords: 17alpha-Hydroxylase deficiency, hyperaldosteronism, congenital adrenal hyperplasia, hypertension, hypokalemia.
INTRODUCCIÓN
La hiperplasia suprarrenal congénita (HSC) es un trastorno causado por diferentes defectos genéticos que afectan a las enzimas involucradas en la esteroidogénesis y biosíntesis del cortisol. La deficiencia de 17alfa-hidroxilasa (17AH) es un trastorno autosómico recesivo que representa alrededor del 1% de todos los casos de HSC con una frecuencia estimada de aproximadamente 1 caso por cada 50.000 individuos1.
La deficiencia de 17AH es causada por alteraciones en el gen que codifica el citocromo P450c171,2. Las alteraciones endocrinas características de esta condición son disminución marcada de la producción de cortisol e hipersecreción compensatoria de la hormona adrenocorticótropa (ACTH), lo que estimula la producción de corticosterona y 11-deoxicorticosterona (11-DOC) en la glándula suprarrenal. Las altas concentraciones de esta última sustancia, que es un mineralocorticoide potente, producen hipertensión, hipokalemia, supresión del eje renina-angiotensina con concentraciones plasmáticas bajas de aldosterona2,3. No obstante, existen informes de concentraciones de aldosterona normales o elevados4. Esta deficiencia también afecta la síntesis de hormonas sexuales en la glándula suprarrenal, lo que provoca virilización defectuosa y alteración del desarrollo sexual 46 XY en varones e infantilismo sexual en las mujeres2. Se presenta un caso de deficiencia de 17alfa-hidroxilasa con hiperaldosteronismo.
CASO CLÍNICO
Se trata de paciente femenina de 16 años de edad quien fue referida a la consulta externa de Endocrinología por presentar amenorrea primaria acompañada de ausencia de caracteres sexuales secundarios. Refería antecedentes de hipertensión de aproximadamente 18 meses de evolución tratada de forma irregular con varios fármacos. Los padres referían antecedentes reproductivos y puberales normales. Negaba antecedentes familiares de enfermedades endocrinas, hipertensión u otras enfermedades crónicas.
Al examen físico, la paciente tenía talla de 1,56 metros y peso de 51 kilogramos (índice de masa corporal 20,9 Kg/m2) y presión arterial de 160 / 95 mm de Hg. En la exploración física fue evidente falta de desarrollo mamario (estadio I de Tanner). Los genitales externos tenían apariencia normal, sin clitoromegalia pero con ausencia de vello púbico y axilar. No se encontraron alteraciones de otros órganos o sistemas durante la evaluación.
Las pruebas de laboratorio mostraron valores séricos de glicemia, pruebas hepáticas, examen de orina y funcionalismo renal normales con valor de potasio bajo (3,0 mEq/L; valor normal [VN] 3,5 - 5,2 mEq/L) y sodio normal (138 mEq/L; VN: 135 - 145 mEq/L). Los resultados de las pruebas hormonales fueron: ACTH a las 8 a.m. de 106 pg/mL (VN: 10 - 46 pg/mL), cortisol a las 8 a.m., 1,75 µg/mL (VN: 6,2 - 19,4 µg/mL), sulfato de dehidroepiandrosterona (S-DHEA) 6,3 µg/mL (VN: 25 - 250 µg/mL), 11-DOC, 250 ng/mL (VN: 4 - 12 ng/mL), 17alfa-hidroxiprogesterona, 4,63 ng/dL (VN: 31 - 217 ng/dL), testosterona total, 5,3 ng/dL (VN: 10 – 70 ng/dL), hormona luteinizante (LH), 34 mUI/mL (VN: fase folicular 2 - 11 mUI/mL), hormona foliculoestimulante (FSH), 89 mUI/mL (VN: fase folicular 4 - 9 mUI/mL) y estradiol, 20 pg/mL (VN: fase folicular 27 - 122 pg/mL). La prueba de estimulación de ACTH no mostró modificaciones tras la administración endovenosa de Synacthen 0,25 mg comparado con la concentración basal. En vista de los hallazgos se decidió realizar mediciones de aldosterona que mostraron valores elevados (385,4 pg/mL; VN 20 - 220 pg/ml) acompañado de disminución de la actividad de la renina plasmática en posición vertical (menor de 0,2 ng/mL/h; VN: 0,2 - 6 mg/mL/h).
La ecografía abdomino-pélvica mostró útero hipoplásico, rudimentario, con endometrio fino y ovarios bilaterales, multiquísticos y pequeños. Las imágenes de tomografía computarizada abdomino-pélvica confirmaron los hallazgos pélvicos y mostraron glándulas suprarrenales sin signos de hiperplasias ni tumoraciones. El electrocardiograma mostró ritmo sinusal y cambios asociados a la hipokalemia, incluyendo intervalo QT prolongado.
Al reexaminar los antecedentes, sintomatología (hipertensión hipocalémica y ausencia de caracteres sexuales secundarios), resultados de laboratorio (concentraciones bajas de cortisol y elevadas de ACTH y FSH) y los resultados de los estudios por imágenes, fue evidente la posibilidad diagnóstica de deficiencia de 17AH. El análisis genético encontró cariotipo normal (46,XX) con mutación heterocigota en el exón 6 del gen CYP17A1 (c.985-987TAC>AA y c.1118A>T). Estos resultados confirmaron el diagnóstico.
La paciente fue tratada inicialmente con hidrocortisona oral (25 miligramos divididos en 3 dosis diarias) y suplementación con cloruro de potasio (1200 mg/día), con mejoría de los valores de presión arterial (110/80 mm de Hg) y concentraciones séricas de potasio (4,1 mEq/L). También fueron indicados ciclos con estrógenos conjugados (0,625 mg/día) para inducir la aparición de los caracteres sexuales secundarios. El suplemento de potasio fue suspendido luego del primer mes de tratamiento con corticosteroides.
En la siguiente consulta (3 meses) se encontró desarrollo mamario incipiente, concentraciones séricas de potasio normales, con aumento de la presión arterial (180/100 mm de Hg), por lo cual la dosis de hidrocortisona fue aumentada a 30 mg/día junto con amlodipino 10 mg/día. Luego de 3 meses de este tratamiento los valores de presión arterial y las concentraciones séricas de potasio permanecieron dentro de límites normales. Los valores de aldosterona (194 pg/mL) y la actividad de renina plasmática (0,76 ng/mL/h) también estaban dentro de límites normales. La exploración física mostró desarrollo mamario con escasa producción de vello púbico y axilar.
DISCUSIÓN
La deficiencia de 17AH es la forma menos común de HSC, está caracterizada por hipertensión, hipokalemia y amenorrea primaria en mujeres, cuyo primer caso fue descrito en 1966.. Esta condición puede permanecer asintomática y no ser diagnosticada hasta la edad adulta temprana. Hasta la fecha se han descrito alrededor de 150 casos en la literatura4.
La mayoría de las mutaciones conocidas en estos casos parecen ser mutaciones de novo del citocromo P450 17A1, localizada en el cromosoma 10q24.3. Hasta la fecha, se han descrito más de 50 mutaciones que afectan las actividades de la hidroxilasa y/o liasa en ciertos grupos étnicos de forma predominante6,7. Las funciones tanto de la 17AH como la 17,20-liasa, son esenciales para la biosíntesis de cortisol, andrógenos y estrógenos..
La deficiencia de la enzima 17AH bloquea la síntesis de cortisol y hormonas sexuales en la glándula suprarrenal. En estos casos, los precursores de esteroides previos al bloqueo enzimático son derivados a la vía de la progesterona, para luego llevar a una sobreproducción en 11-DOC y corticosterona por una vía enzimática diferente a la 17AH6,7. Además, este bloqueo aumenta la síntesis de la vía de los mineralocorticoides, llevando a alteraciones de las concentraciones de 17-hidroxipregnolona, 17-hidroxiprogesterona, 11-deoxicortisol, cortisol, DHEA-S, androstenediona, testosterona y a sobreproducción de 17-deoxiesteroides por la corteza suprarrenal, incluyendo 18-hidroxicorticosterona (figura 1). Las concentraciones de la FSH y LH, como fue demostrado en la paciente de este caso, generalmente están elevadas por la falta de retroalimentación negativa de los esteroides sexuales..
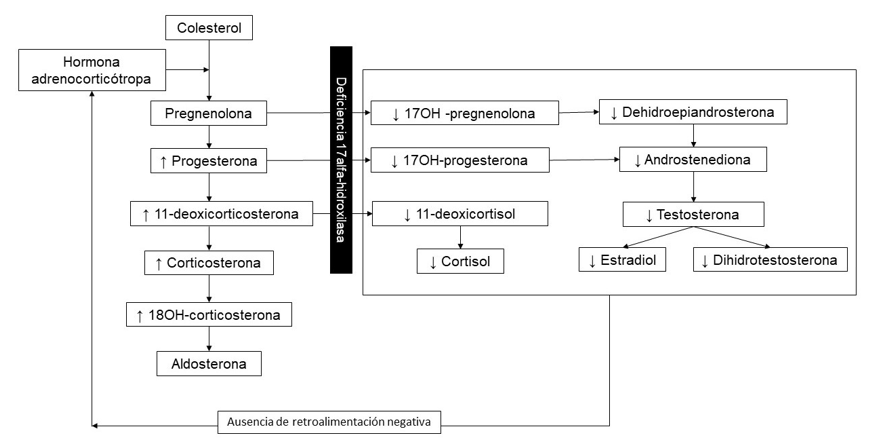
Fig. 1
Alteración de las vías de la esteroidogénesis en la deficiencia de 17alfa-hidroxilasa.
El eje renina-angiotensina-aldosterona es independiente del eje hipotálamo-hipófisis-suprarrenal y tiene regulaciones e interacciones complicadas6,7. Los mineralocorticoides resultantes del bloqueo actúan sobre los túbulos renales produciendo la excreción de potasio y reabsorción de sodio - agua, lo que conduce a aumento del volumen plasmático, hipertensión e hipokalemia. En la mayoría de los casos, la aldosterona esta suprimida por las altas concentraciones de mineralocorticoides que inducen aumento de la actividad de la corticosterona metil oxidasa2,6.
El presente caso es una variante de la deficiencia de 17AH clásica con hiperaldosteronismo y disminución de la actividad de la renina. Existen informes de alrededor de 20 casos con valores normales y otros 20 casos con concentraciones elevadas de aldosterona8. Algunos investigadores han propuesto que mientras mayor es la deficiencia enzimática, mayores son las concentraciones de aldosterona9, sin embargo, esta hipótesis no ha sido validada8. Por otra parte, el mecanismo que produce los bajos valores de actividad de renina aún debe ser identificado.
El diagnóstico de la deficiencia de 17AH está basado en las manifestaciones clínicas, hallazgos bioquímicos y moleculares. Como en este caso, la condición debe sospecharse en sujetos jóvenes con hipertensión primaria, hipokalemia y ausencia de los caracteres sexuales secundarios. En la mayoría de los casos el cariotipo es normal, pero el retraso puberal es más común en las mujeres. Las pruebas de laboratorio suelen mostrar concentraciones séricas elevadas de ACTH y FSH junto a concentraciones bajas de testosterona y estradiol. La progesterona es uno de los sustratos que puede acumularse en la deficiencia de 17AH, por lo que el aumento de las concentraciones séricas puede ser un marcador diagnóstico útil para esta condición10. Dado que las características clínicas y bioquímicas son diversas, dependiendo del tipo de mutación, el análisis genético es fundamental para confirmar el diagnóstico, como se demostró en esta paciente11.
La supresión de la producción de hormonas sexuales en la deficiencia de 17AH produce hipogonadismo hipergonadotrópico con diferentes características fenotípicas2,12. El análisis de la actividad enzimática ha demostrado que es necesario más del 25% de la actividad normal para el desarrollo sexual fetal normal de los genitales externos2,5. Los sujetos XX generalmente tienen órganos reproductores femeninos rudimentarios, pero no desarrollan características sexuales secundarias, como vello púbico o del tejido mamario, ni experimentan menarquia. Los individuos XY no tienen útero ni trompas de Falopio porque los testículos producen el factor inhibidor que provoca la regresión del conducto de Müller. Aquellos casos con insuficiencia parcial pueden presentar genitales externos ambiguos o poco virilizados, característicos de la alteración del desarrollo sexual 46,XY3,13.
El tratamiento de la deficiencia de 17AH incluye la administración de terapia de reemplazo con glucocorticoides y hormonas sexuales. El propósito del tratamiento con glucocorticoides es normalizar las concentraciones séricas de 11-DOC y ACTH, suprimiendo el exceso de mineralocorticoides. Pueden utilizarse dexametasona (0,25 - 0,5 mg), prednisona (2 - 4 mg/m2) o hidrocortisona (10 - 15 mg/m2)1,5. En el presente caso se seleccionó la hidrocortisona por su menor efecto mineralocorticoide. La determinación de las concentraciones de ACTH no es útil para evaluar el éxito del tratamiento, ya que la normalización de sus valores puede requerir dosis supra-fisiológicas de glucocorticoides6.
En la mayoría de los casos, el tratamiento con glucocorticoides puede controlar el aumento de la presión arterial, pero en ocasiones la hipertensión puede persistir14. En estos casos puede ser necesario utilizar antagonistas de los mineralocorticoides, como espironolactona. En vista de que el tratamiento de la paciente con glucocorticoides no logró normalizar las cifras de presión arterial, se tomó la decisión de indicarle amlodipino. El tratamiento logró controlar la hipertensión. El uso de bloqueadores de los canales de calcio ha demostrado ser eficaz si la hipertensión persiste a pesar de la resolución de las alteraciones endocrinas de los mineralocorticoides2,15.
La sustitución de hormonas sexuales es fundamental. Como en el presente caso, los estrógenos pueden administrarse en las pacientes con cariotipo 46,XX11. En estos casos el reemplazo consiste en administración secuencial de estrógenos seguidos de progestinas o anticonceptivos orales. El propósito es lograr el desarrollo mamario - uterino y el desarrollo de las características sexuales secundarias. Este tratamiento induciría la hemorragia por deprivación cíclica, previniendo la hiperplasia endometrial. También pueden añadirse testosterona a dosis bajas para contribuir al desarrollo de las características sexuales secundarias1,9. Las pacientes con deficiencia de 17AH no pueden producir ovocitos maduros debido al fracaso del desarrollo folicular secundario al defecto irreversible de la esteroidogénesis4. La gonadectomía profiláctica está indicada en sujetos XY debido al riesgo de cambios malignos. Las gónadas, en estos casos, pueden encontrarse intra-abdominales, en el canal inguinal o en los pliegues labio-escrotales..
CONCLUSIÓN
La deficiencia de 17AH es una forma rara de HSC y los casos que presentan hiperaldosteronismo son aún menos frecuentes. La mayoría de los pacientes presentan los síntomas en el momento de la pubertad, por lo que es difícil el diagnóstico temprano. Es necesario considerar esta condición en pacientes jóvenes con hipertensión, hipokalemia y ausencia de desarrollo sexual secundario. El tratamiento con glucocorticoides y hormonas sexuales debe iniciarse lo más pronto posible para evitar complicaciones.
CONFLICTOS DE INTERÉS
Los autores declaran que no presentan conflictos de interés.
REFERENCIAS BIBLIOGRÁFICAS
1. Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol 2017;165:71-78.
2. Wong SL, Shu SG, Tsai CR. Seventeen alpha-hydroxylase deficiency. J Formos Med Assoc 2006;105:177-181.
3. Ramos N, Lombès M. Résistances aux hormones stéroïdes: physiologie et pathologie: Pathophysiology of Steroid Resistance Syndrome. Ann Endocrinol (Paris) 2016;77 Suppl 1:S1-S10.
4. Britten FL, Ulett KB, Duncan EL, Perry-Keene DA. Primary amenorrhoea with hypertension: undiagnosed 17-α-hydroxylase deficiency. Med J Aust 2013;199:556-558.
5. Wang M, Wang H, Zhao H, Li L, Liu M, Liu F, Meng F, Fan C. Prevalence of CYP17A1 gene mutations in 17α-hydroxylase deficiency in the Chinese Han population. Clin Hypertens 2019;25:23.
6. Won GS, Chiu CY, Tso YC, Jenq SF, Cheng PS, Jap TS. A compound heterozygous mutation in the CYP17 (17alpha-hydroxylase/17,20-lyase) gene in a Chinese subject with congenital adrenal hyperplasia. Metabolism 2007;56:504-507.
7. Kardelen AD, Toksoy G, Baş F, Yavaş Abalı Z, Gençay G, Poyrazoğlu Ş, Bundak R, Altunoğlu U, Avcı Ş, Najaflı A, et al. A rare cause of congenital adrenal hyperplasia: clinical and genetic findings and follow-up characteristics of six patients with 17-hydroxylase deficiency including two novel mutations. J Clin Res Pediatr Endocrinol 2018;10:206-215.
8. Zhu Z, Ni S, Gu W. Clinical characteristics and mutation analysis of two Chinese children with 17a-hydroxylase/17,20-lyase deficiency. Int J Clin Exp Med 2015;8:19132-19137.
9. Lee SJ, Song JE, Hwang S, Lee JY, Park HS, Han S, Rhee Y. Untreated congenital adrenal hyperplasia with 17-α hydroxylase/17,20-lyase deficiency presenting as massive adrenocortical tumor. Endocrinol Metab (Seoul) 2015;30:408-413.
10. Zhou Y, Xue X, Shi P, Lu Q, Lv S. Multidisciplinary team management of 46,XY 17α-hydroxylase deficiency: a case report and literature review. J Int Med Res 2021;49:300060521993965.
11. Li H, Qiao J, Guo H. 17-alpha-hydroxylase deficiency: a case report with clinical and molecular analysis. Gynecol Endocrinol 2010;26:521-523.
12. Kim YM, Kang M, Choi JH, Lee BH, Kim GH, Ohn JH, Kim SY, Park MS, Yoo HW. A review of the literature on common CYP17A1 mutations in adults with 17-hydroxylase/17,20-lyase deficiency, a case series of such mutations among Koreans and functional characteristics of a novel mutation. Metabolism 2014;63:42-49.
13. Unal E, Yıldırım R, Taş FF, Tekin S, Ceylaner S, Haspolat YK. A rare cause of delayed puberty in two cases with 46,XX and 46,XY karyotype: 17 alpha-hydroxylase deficiency due to a novel variant in CYP17A1 gene. Gynecol Endocrinol 2020;36:739-742.
14. Sun X, Li M, Wang Y, Bi Y. Two novel heterozygous mutations in the CYP17A1 gene in a Chinese patient with 17α-hydroxylase 17,20-lyase deficiency. Discov Med 2018;26:243-249.
15. de Silva T, Cosentino G, Ganji S, Riera-Gonzalez A, Hsia DS. Endocrine causes of hypertension. Curr Hypertens Rep. 2020;22:97.
Notas de autor
Dirigir correspondencia a: Eduardo Reyna-Villasmil. Email: sippenbauch@gmail.com