Casos clínicos
COEXISTENCIA DE CRANEOFARINGIOMA Y MENINGIOMA
COEXISTENCE OF CRANIOPHARYNGIOMA AND MENINGIOMA
COEXISTENCIA DE CRANEOFARINGIOMA Y MENINGIOMA
Revista Venezolana de Endocrinología y Metabolismo, vol. 20, núm. 2, pp. 113-119, 2022
Sociedad Venezolana de Endocrinología y Metabolismo

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 3.0 Internacional.
Recepción: 01 Diciembre 2021
Aprobación: 01 Abril 2022
Resumen:
Objetivo: Describir la coexistencia de dos tipos de tumores diferentes, craneofaringioma y meningioma, sus implicaciones clínicas y tratamiento.
Caso Clínico: Masculino de 68 años de edad quien presenta desde hace 3 meses pérdida de la memoria reciente, lenguaje incoherente, alucinaciones visuales y auditivas, astenia, adinamia y somnolencia. Valores de laboratorio: glucemia 67 mg/dL, sodio: 134 mmol/L y cortisol 8 am 8,2 μg/dL. En la resonancia magnética nuclear (RMN) se evidencia lesión ocupante de espacio de componente mixto, supraselar con extensión intraventricular, compatible con craneofaringioma, y un meningioma de la región sellar con preservación de la glándula hipofisaria. El tratamiento quirúrgico se basó en la colocación de catéter guiado por esterotaxia conectado a reservorio de Ommaya para drenar el componente quístico de la lesión. El meningioma fue manejado de forma conservadora. Se evidenció una franca mejoría de la conducta y de la memoria reciente.
Conclusión: El craneofaringioma combinado con meningioma es un tumor de colisión extremadamente raro, encontrando en la literatura solo 8 reportes de casos; se presenta a paciente masculino de 68 años con diagnóstico de craneofaringioma, de componente suprasellar complicado con insuficiencia adrenal, evidenciándose de forma incidental meningioma en el estudio de RMN cerebral. El tratamiento, previo a reemplazo hormonal con glucocorticoides, consistió en el drenaje del componente líquido de la lesión mediante la colocación de catéter con reservorio de Ommaya, sin complicaciones y con buena evolución en el postoperatorio.
Palabras clave: Craneofaringioma del adulto, meningioma, insuficiencia adrenal, pérdida de memoria, tumor de colisión, tumores coexistentes.
Abstract:
Objective: To describe the coexistence of two different types of tumors in the same patient, craniopharyngioma and meningioma, its clinical management and treatment.
Clinical Case: A 68 year-old male who has presented recent memory loss for 3 months, incoherent language, visual and auditory hallucinations, asthenia, adynamia and drowsiness. Laboratory values: blood glucose 67 mg/dL, sodium: 134 mmol/L and cortisol 8 am 8.2 μg/dL (5-23 μg/dL). The magnetic resonance imaging (MRI) shows a space-occupying lesion of mixed component, suprasellar with intraventricular extension, compatible with craniopharyngioma, and a meningioma of the sellar region with preservation of the pituitary gland. Surgical treatment was based on the placement of a stereotaxis-guided catheter connected to the Ommaya reservoir to drain the cystic component of the lesion. The meningioma was managed conservatively. A clear improvement in behavior and recent memory was evidenced.
Conclusion: Craniopharyngioma combined with meningioma is an extremely rare collision tumor, with only 8 case reports found in the literature; a 68-year-old male patient is presented with a diagnosis of craniopharyngioma, with a suprasellar component complicated by adrenal insufficiency, incidentally showing meningioma in the brain MRI study. The treatment, prior to hormone replacement with glucocorticoids, consisted of draining the liquid component of the lesion by placing an Ommaya reservoir catheter, without complications and with good postoperative evolution.
Keywords: Adult craniopharyngioma, meningioma, adrenal insufficiency, memory loss, limbic system, collision tumor, coexistent tumors.
INTRODUCCIÓN
La región sellar y parasellar es anatómicamente compleja y puede ser el sitio de varios tumores diferentes a los adenomas hipofisarios en el 10-15% de los casos1, siendo el craneofaringioma y el meningioma los tumores benignos más comunes. El craneofaringioma es una malformación embrionaria poco frecuente, de bajo grado histológico, de crecimiento lento, que se origina de restos celulares de la bolsa de Rathke a lo largo de una línea que va desde la nasofaringe hasta el diencéfalo2-7. Los meningiomas surgen de la aracnoides, suelen ser neoplasias benignas de crecimiento lento, representan el tumor intracraneal benigno más frecuente1,8,9. La presencia de al menos dos tipos de tumores en una posición craneal se define como tumor coexistente, lo cual es poco común en neurocirugía10. A continuación, se presenta un caso con diagnóstico de craneofaringioma y meningioma coexistente.
CASO CLÍNICO
Paciente masculino de 68 años de edad quien acude por presentar pérdida de la memoria reciente, lenguaje incoherente, alucinaciones visuales y auditivas, astenia, adinamia y somnolencia de aproximadamente 3 meses de evolución. Antecedente de hipertensión arterial e hiperinsulinismo, en tratamiento con losartan 50 mg y metformina 500 mg vía oral, orden diaria. En el interrogatorio dirigido refiere estreñimiento crónico y episodio de cefalea intensa desde hace 3 meses, que no cede con analgésicos. Al examen físico el paciente luce en buenas condiciones generales con signos vitales dentro de la normalidad, normopeso dado por un índice de masa corporal de 22 kg/m2. Tiroides apenas palpable. Neurológicamente, consciente, desorientado en tiempo y espacio, obedece órdenes sencillas, pérdida de la memoria reciente y trastornos de conducta con crisis de agresividad. El resto de la exploración física no mostró hallazgos patológicos.
Exámenes de laboratorio: glucemia en 67 mg/dL (70-100 mg/dL), nitrógeno ureico: 8,9 mg/dL (7-25 mg/dL), creatinina: 1,05 mg/dL (0,7-1,2 mg/dL), sodio: 134 mmol/L (135-145 mmol/L), potasio: 3,8 mmol/L (3,5-5,1 mmol/L), cloro 101 mmol/L (98-106 mmol/L), T4L 0,9 ng/dL (0,8-2,2 ng/dL) y cortisol 8 am de 8,2 μg/dL (5-23 μg/dL).
En el estudio de resonancia magnética nuclear (RMN) se evidencia una lesión ocupante de espacio (LOE) mixta, sólida y quística, supraselar con extensión intraventricular, sin hidrocefalia, compatible con un craneofaringioma y un meningioma (Ver figura 1 A y B). En el estudio se puede evidenciar como la lesión desplaza el techo del tercer ventrículo o el piso del ventrículo lateral, el cual forma parte del sistema límbico donde se encuentra parte de los circuitos de la memoria reciente, por lo que su compresión explica la pérdida de memoria reciente con la cual se presenta el paciente.
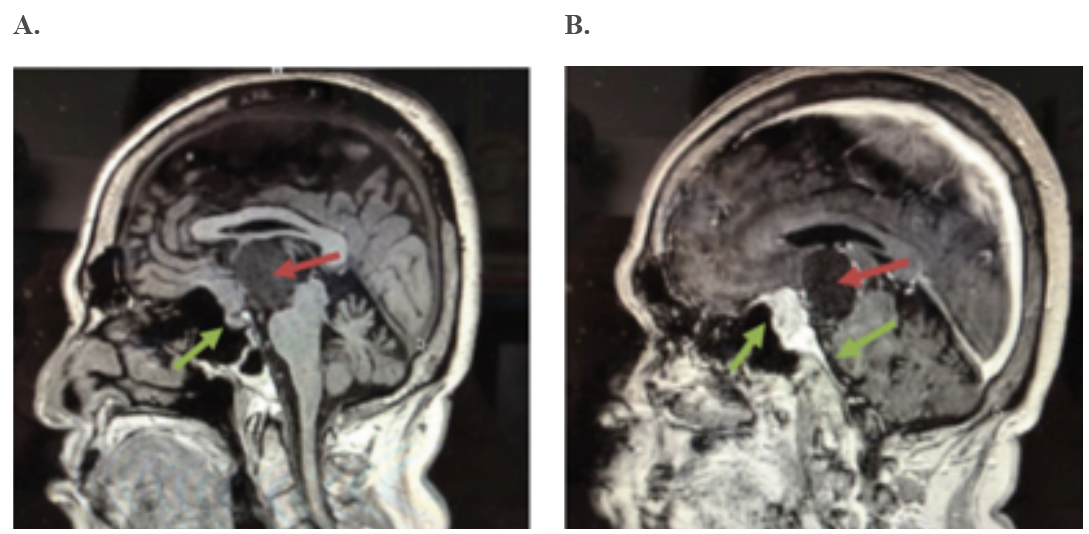
Figura 1
RMN en T1, corte sagital. A. Pre-contraste: se aprecia lesión hipodensa heterogénea supraselar con extensión intraventricular (flecha roja). Se observa la glándula hipófisis indemne en sus dos porciones (flecha verde); la porción hiperintensa corresponde a la neurohipófisis). B. Post contraste: Lesión heterogénea quística (flecha roja) sugestiva de craneofaringioma y lesión hiperdensa que capta contraste evidenciándose cola dural tanto en el plano esfenoidal como en el clivus en relación a meningioma (flechas verdes).
Se plantean los siguientes diagnósticos: 1. LOE mixto supraselar con extensión intraventricular compatible con craneofaringioma 2. Meningioma de la región sellar. 3. Insuficiencia adrenal (IA) secundaria o terciaria, en vista de presentar valores de glucemia en 67 mg/dL, Na 134 mmol/L, niveles de cortisol < 10 μg/dL con clínica de astenia y adinamia. El paciente no se pudo realizar la prueba de estimulación con corticotropina para confirmar el diagnóstico de insuficiencia adrenal. El diagnóstico se realizó en base a la clinica del paciente y hallazgos imagenológicos, pudiéndose plantear IA secundaria o terciaria, ya que la lesión tiene un componente supraselar y la glándula hipófisis se encuentra indemne. Se indicó reemplazo con glucocorticoides en el preoperatorio.
El tratamiento quirúrgico se basó en la colocación de catéter guiado por esterotaxia conectado a reservorio de Ommaya para drenar el componente quístico de la lesión, por ser este el de mayor volumen. El objetivo fue la descompresión de las estructuras neurológicas con un bajo riesgo quirúrgico. El meningioma no fue resecado, ya que su ubicación no explica la clínica del paciente. No hubo diabetes insípida central ni complicaciones en el posoperatorio. La biopsia en los extendidos citológicos de la lesión evidenció una sustancia de fondo espesa, homogénea de color rojizo, de carácter lipídico, presencia de elementos cristaloides y escasos hematíes. Hallazgos compatibles con craneofaringioma.
La evolución del paciente fue satisfactoria evidenciándose una franca mejoría de la conducta y de la memoria reciente en un plazo de 6 semanas. En la RMN control a los 3 meses de la cirugía se evidenció una franca reducción de la lesión quística, que se ha mantenido en el tiempo, demostrándose descompresión de las estructuras neurológicas. Se observa el meningioma del mismo tamaño al estudio preoperatorio (ver figura 2).
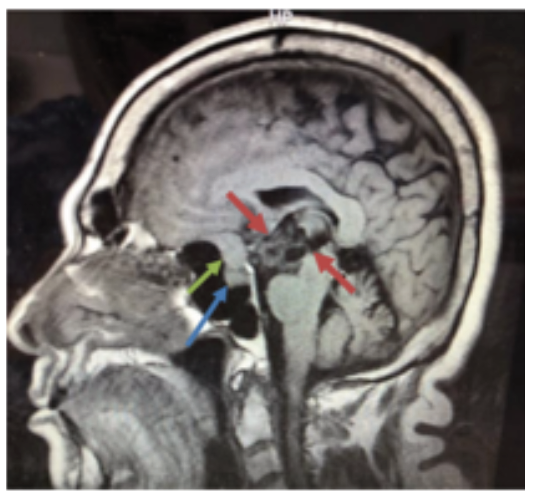
Figura 2
RMN cerebral posterior al drenaje del componente quístico del craneofaringioma. T1, corte sagital: se aprecia lesión heterogénea, que muestra franca disminución de su volumen en comparación al preoperatorio. Restos del craneofaringioma y su cápsula (flechas rojas). Meningioma (flecha verde) e Hipófisis (flecha azul)
DISCUSIÓN
El craneofaringioma es una malformación embrionaria que se origina de restos celulares de la bolsa de Rathke a lo largo de una línea que va desde la nasofaringe hasta el diencéfalo2-7. La mayoría de estos tumores epiteliales ocurren en la región sellar, para y suprasellar. Representan entre el 2% y el 5% de todos los tumores intracraneales primarios con una tasa de incidencia general de 0,5 a 2 casos por millón por año2. Se ha demostrado una distribución por edades bimodal, con tasas de incidencia máximas en niños de 5 a 14 años y adultos de 50 a 74 años4. En los estudios basados en la población, no se han observado diferencias de género4,6.
A pesar de ser tumores benignos, a menudo infiltran estructuras adyacentes como la hipófisis, el hipotálamo, nervios ópticos, vasos sanguíneos o el tercer ventrículo, lo que provoca una morbilidad y mortalidad significativas3. Los pacientes con craneofaringioma de inicio en la edad adulta tienen una mortalidad mucho más alta en comparación con la población general3.
Los craneofaringiomas se dividen en dos subtipos histológicos: el tipo adamantinomatoso y el papilar2-4. El tipo adamantinomatoso puede desarrollarse en todas las edades, mientras que el tipo papilar ocurre exclusivamente en adultos, rara vez se presenta con calcificaciones, generalmente está bien circunscrito y en comparación con el tipo adamantinomatoso, la infiltración tumoral del tejido circundante es menos frecuente3. Pueden surgir en cualquier parte del canal craneofaríngeo, pero la mayoría de ellos ocurren en la región sellar/parasellar2-4. El 94-95% tiene un componente suprasellar, siendo entre el 20-41% puramente suprasellar y entre el 53-75% tanto suprasellar como intrasellar2,4. Los puramente intrasellares representan la variedad menos común2,4.
La proximidad y el efecto de masa de los craneofaringiomas sobre las estructuras vitales del cerebro como las vías visuales, el parénquima cerebral, el sistema ventricular, los vasos sanguíneos principales y el eje hipotálamo-hipofisario predisponen a los pacientes a múltiples manifestaciones clínicas, cuya gravedad va a depender de la ubicación, el tamaño y el potencial de crecimiento del tumor2,3. Los síntomas son con frecuencia inespecíficos, desarrollándose gradualmente, lo que contribuye a un retraso en el diagnóstico de 1-2 años3,6.
En adultos, los síntomas clínicos más comunes son el déficit del campo visual y los signos de hipopituitarismo3. En alrededor del 40-84% de los pacientes la extensión del tumor suprasellar comprime el quiasma óptico ocasionando defectos del campo visual3,6, que suelen presentarse como hemianopsia bitemporal hasta en el 49% de los casos2. La cefalea se ha reportado en el 56% de los casos, en mujeres irregularidades menstruales en el 57%, pérdida de energía entre el 32 al 48%, náuseas y vómitos en el 26%, letargo en el 26% y aumento de peso entre el 13 al 15% de los casos6. Síntomas menos frecuentes como cambios en la personalidad y deterioro cognitivo, como el descrito en el caso clínico, se han reportado en el 8 al 17% de los casos respectivamente3, especialmente en adultos de mediana edad2.
Entre el 40 al 87% de los pacientes con craneofaringioma de inicio en la edad adulta, presentan al momento del diagnóstico por lo menos un déficit hormonal4, causado por alteraciones de los ejes hipotalámico-hipofisario. Un resumen de los resultados de varios estudios, en los que se han adoptado diferentes pruebas y criterios diagnósticos, muestra que el déficit de hormona de crecimiento (HC) está presente en el 35-95% de los pacientes evaluados, el déficit de gonadotropinas (FSH / LH) en el 38-82%, deficiencia de ACTH entre el 21 a 62%, deficiencia de TSH en 21 a 42% y diabetes insípida en 6 a 38% de los casos2.
La evaluación de la afectación hipotalámica en el craneofaringioma es importante para estimar el pronóstico y la calidad de vida a largo plazo6. La afectación tumoral inicial del piso del tercer ventrículo, los cuerpos mamarios y/o el hipotálamo posterior en las imágenes se asocia con un peor pronóstico a largo plazo debido a la obesidad hipotalámica, independientemente de las estrategias de tratamiento elegidas6.
Para la evaluación del eje hipotálamo hipófisis adrenal se solicita cortisol serico a las 8 am. Un valor < 3 μg/dL es indicativo de insuficiencia adrenal y un valor >15 μg/dL excluye el diagnóstico7. Si los valores de cortisol están entre 3 y 15 μg/dL, la Sociedad Americana de Endocrinología recomienda realizar el test de estimulación con corticotropina para confirmar el diagnóstico de insuficiencia adrenal7. Este test evalúa la reserva adrenal de cortisol basado en la premisa de que la deficiencia crónica de ACTH resulta en atrofia de la corteza suprarenal y ésta como consecuencia no responde a un solo pulso de corticotropina8. El test de estimulación estándar con corticotropina consiste en administrar 250 μg de corticotropina (Cortrosyn®) IM o IV y medir niveles de cortisol a los 30 y/o 60 min. Para excluir insuficiencia adrenal el cortisol debe estar a los 30 o 60 min ≥18,1-20 μg/dl, dependiendo del ensayo realizado7. Actualmente, la Sociedad Americana de Endocrinología recomienda que el límite de cortisol sérico a los 30-60 minutos sea > 14 a 15 μg/dL, para disminuir los falsos positivos9.
En el caso presentado no se pudo realizar el test de estimulación con corticotropina para confirmar el diagnóstico de insuficiencia adrenal secundaria, éste diagnóstico fue realizado en base a los valores bajos de glucemia y sodio, clínica de adinamia y astenia, y a la presencia de un LOE mixto suprasellar con extensión intraventricular compatible con craneofaringioma. Por ese motivo se decidió reemplazar con glucocorticoides previo a la cirugía, para evitar crisis de insuficiencia adrenal aguda durante el acto quirúrgico y en el postoperatorio.
Tanto la tomografía computarizada como la resonancia magnética demuestran que el craneofaringioma es típicamente un tumor quístico de la región intra y/o suprasellar2-5. La tomografía computarizada es la herramienta que permite detectar o excluir calcificaciones con mayor facilidad en el tejido del craneofaringioma2-5, que se encuentra en aproximadamente el 90% de estos tumores4. La intensidad de la señal del craneofaringioma en la resonancia magnética es muy variable porque depende de la concentración de proteínas en el líquido quístico2,4. Las porciones sólidas del tumor y las membranas del quiste aparecen isointensas en la resonancia magnética ponderada en T1, a menudo con una estructura levemente heterogénea4. La combinación de componentes tumorales sólidos, quísticos y calcificaciones es un hallazgo radiológico característico para el diagnóstico4.
La estrategia terapéutica óptima del craneofaringioma es controversial3,6,10. No existen pautas basadas en la evidencia, ni un consenso claro para el manejo de los craneofaringiomas primarios o recurrentes en adultos3. El tratamiento radical con resección completa del tumor y una cura potencial debe equilibrarse con un enfoque más conservador para evitar la morbilidad y mortalidad sustancial asociada al tratamiento3. Por lo tanto, el tratamiento óptimo siempre debe individualizarse teniendo en cuenta los síntomas del paciente, la edad, la localización del tumor y la extensión2. Las complicaciones endocrinas y metabólicas deben tratarse antes de la cirugía o de la radioterapia. Esto es especialmente importante en caso de insuficiencia adrenal, tiroidea o diabetes insípida3.
El tratamiento quirúrgico de estas lesiones sigue siendo uno de los más desafiantes para los neurocirujanos debido a sus relaciones topográficas complejas con estructuras neurales y vasculares cruciales como el quiasma óptico, el hipotálamo, el tercer ventrículo y los vasos del polígono de Willis6. Tradicionalmente, el objetivo de la cirugía había sido la resección total o casi total (>95%) del tumor, principalmente por craneotomía, sin embargo, los efectos secundarios derivados de esta estrategia, como la lesión hipotalámica, el deterioro cognitivo y el empeoramiento secundario de la calidad de vida, apoyan el empleo de una resección más limitada seguida de radioterapia10. No existe un paradigma de mejor tratamiento y el alcance de la resección quirúrgica o la terapia adyuvante debe considerarse de forma individual6.
El 90% de los craneofaringiomas tienen un componente quístico, que en ocasiones constituye el principal componente del tumor y requiere un tratamiento específico10. Las lesiones con componente quístico importante que no son susceptibles de exéresis completa pueden ser tratadas por punción por esterotaxia o mediante aspiración intermitente a través de reservorio tipo Ommaya10, tal como se realizó en este caso.
Los meningiomas surgen de las células aracnoideas, suelen ser neoplasias benignas de crecimiento lento1,11. Aproximadamente el 15% de ellos se desarrollan a partir de la región parasellar, en el seno cavernoso, el tubérculo de la silla turca, el diafragma, el dorso de la silla, las apófisis clinoides anterior y posterior y el clivus12. Los meningiomas son más prevalentes en mujeres y su incidencia aumenta con la edad11,12.
La presentación clínica de los meningiomas suele ser inespecífica, la localización y compresión de las estructuras vasculares y cerebrales adyacentes puede producir déficits neurológicos focales11. Los síntomas que se observan comúnmente son los siguientes: cefalea en el 33,3 a 36,7%, déficit focal de pares craneales entre el 28,8 y el 31,3%, convulsiones en el 16,9 a 24,6%, cambio cognitivo en el 14,4%, debilidad en el 11,1%, vértigo/mareo en el 9,8%, ataxia/cambio de marcha en el 6,3%, dolor/cambio sensorial en el 5,6%, proptosis en el 2,1%, síncope en el 1,0% y asintomático hasta en el 9,4% de los casos11. Los meningiomas intracraneales diagnosticados de forma incidental ocurren hasta en 30% de los casos13.
El diagnóstico se basa principalmente en la evaluación neurológica y radiológica, aunque la función hipofisaria también puede verse afectada1,11,12. Imagenológicamente, los meningiomas suelen aparecer como masas homogéneas, hiper o isodensas en comparación con la sustancia gris en la tomografía computarizada, presentando calcificación difusa en muchos casos. Son isointensos u ocasionalmente hipointensos en la resonancia magnética ponderada en T1, realzados de manera brillante y homogénea después del gadolinio y variables en la resonancia magnética ponderada en T2. La típica cola de duramadre (un realce de contraste dural adyacente) se observa con frecuencia en las imágenes, aunque no se considera una característica patognomónica específica del meningioma12.
El tratamiento depende de las manifestaciones clínicas, la edad del paciente y su tamaño1. Los meningiomas asintomáticos pueden tratarse de forma conservadora mediante observación, reservándose la cirugía para lesiones sintomáticas o en crecimiento1. En una revisión sistemática y meta-análisis que incluyó 20 estudios con 2130 pacientes, las estrategias de manejo inicial fueron: cirugía en 27,3%, radiocirugía estereotáctica en 22%, monitorización activa en el 50,7% con un seguimiento de 49,5 meses, reportando progresión clínica y radiológica en el 22% de los casos13. Aquellos meningiomas mayores de 3 cm o con edema peritumoral tienen mayor riesgo de progresión y deben ser monitorizados frecuentemente en los primeros 5 años del diagnóstico13.
La presencia de al menos dos tipos de tumores en una posición craneal se define como tumor coexistente14. El craneofaringioma combinado con meningioma es un tumor de colisión extremadamente raro, desde 1967 hasta el 2020 solo se habían reportado 8 casos similares14. Los 2 tumores pueden contribuir mutuamente a la patogenia del otro y puede estar asociada a los siguientes aspectos14: 1. Dos tumores se produjeron en la misma posición o en ubicaciones adyacentes; 2. Los tumores ocurrieron en la misma posición de los diferentes tejidos debido a la radiación, exposición química y trauma; 3. El tumor craneal fue inducido por diferentes tumores derivados de tejido en el parénquima cerebral periférico o tejidos meníngeos; 4. La estructura embrionaria residual finalmente se desarrolló en diferentes tumores derivados de tejidos.
CONCLUSIÓN
Al ser el craneofaringioma combinado con meningioma un tumor de colisión extremadamente raro, encontrando en la literatura sólo 8 reportes de casos, se presenta a paciente masculino de 68 años con diagnóstico de craneofaringioma, predominantemente quístico, de componente suprasellar complicado con insuficiencia adrenal secundaria, evidenciándose de forma incidental meningioma en el estudio de RMN cerebral. El tratamiento, previo a reemplazo hormonal con glucocorticoides, consistió en el drenaje del componente líquido de la lesión mediante la colocación de catéter con reservorio de Ommaya guiado por estereotaxia, sin complicaciones y con buena evolución en el postoperatorio. El diagnóstico del meningioma fue incidental, como se describe hasta en 30% de los casos, de curso asintomático, manejado de forma conservadora.
CONFLICTOS DE INTERÉS
Los autores declaran que no presentan conflictos de interés.
REFERENCIAS BIBLIOGRÁFICAS
1. Syro L, Rotondo F, Moshkin O, Kovacs K. Nonpituitary sellar masses. En: Sholmo Melmed. The Pituitary. Fourth Edition. 2017pp. 631.
2. Karavitaki N, Cudlip S, Adams C, Wass J. Craniopharyngiomas. Endocr Rev 2006;27:371-397. DOI: 10.1210/er.2006-0002
3. Zoicas F, Schofl C. Craniopharyngioma in adults. Front Endocrinol 2012;3:46. DOI: 10.3389/fendo.2012.00046
4. Muller H. Craneopharyngioma. Endocr Rev 2014;35:513-543. DOI: 10.1210/er.2013-1115
5. Muller H. The diagnosis and treatment of craniopharyngioma. Neuroendocrinology 2020;110:753-766. DOI: 10.1159/000504512
6. Jensterle M, Jazbinsek S, Bosnjak R, Popovic M, Zadravec L, Vipotnik T, Faganel B, Kotnik P. Advances in the management of craniopharyngioma in children and adults. Radiol Oncol 2019;53:388-396. doi: 10.2478/raon-2019-0036
7. Fleseriu M, Hashim I, Karavitaki N, Melmed S, Hassan Murad M, Salvatori R, Samuels MH. Hormonal replacement in hypopituitarism in adults: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016;101:3888-3921. doi: 10.1210/jc.2016-2118.
8. Toogod A, Stewart P. Hypopituitarism: clinical features, diagnosis, and management. Endocrinol Metab Clin North Am 2008;37:2352261. doi: 10.1016/j.ecl.2007.10.004.
9. Javorsky BR, Raff H, Carroll TB, Algeciras-Schimnich A, Jit Singh R, Colón-Franco LM, Findling JW. New cutoffs for the biochemical diagnosis of adrenal insufficiency after acth stimulation using specific cortisol assays. J Endocr Soc 2021;5. doi: 10.1210/jendso/bvab022.
10. Venegas E, Blanco C, Martin T, Soto A. Guía práctica del manejo y tratamiento de los craneofaringiomas y otras lesiones paraselares. Endocrinol Nutr 2015;62:e1-e13. https://doi.org/10.1016/j.endonu.2014.05.005.
11. Ogasawara C, Philbrick B, Adamson C. Meningioma: a review of epidemiology, pathology, diagnosis, treatment, and future directions. Biomedicines 2021;9:319. https://doi.org/10.3390/biomedicines9030319.
12. Gatto F, Perez-Rivas LG, Olarescu NC, Khandeva P, Chachlaki K, Trivellin G, Gahete MD, Cuny T, on behalf of the ENEA Young Researchers Committee (EYRC). Diagnosis and treatment of parasellar lesions. Neuroendocrinology 2020;110:728-739. DOI: 10.1159/000506905
13. Islim AI, Mohan M, Moon RDC, Srikandarajah N, Mills SJ, Brodbelt AR, Jenkinson MD. Incidental intracranial meningiomas: a systematic review and meta-analysis of prognostic factors and outcomes. J Neurooncol 2019;142:211-221. doi: 10.1007/s11060-019-03104-3.
14. Liu G, Su L, Xiang Y, Liu Y, Zhang S. Coexistence of craniopharyngioma and meningioma: Two rare cases and literature review. Medicine 2020;99:50:e23183. http://dx.doi.org/10.1097/MD.0000000000023183.
Notas de autor
Dirigir correspondencia a: Ana Cristina Haiek. Email: haiekana@gmail.com