Comunicaciones Breves
TUMOR NEURO-ENDOCRINO DEL PÁNCREAS DE CÉLULAS INSULARES NO FUNCIONANTE
TUMOR NEURO-ENDOCRINO DEL PÁNCREAS DE CÉLULAS INSULARES NO FUNCIONANTE
Revista Venezolana de Oncología, vol. 29, núm. 1, pp. 52-58, 2017
Sociedad Venezolana de Oncología

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 4.0 Internacional.
Recepción: 19 Julio 2016
Revisado: 15 Septiembre 2016
Aprobación: 12 Octubre 2016
Resumen: Los tumores neuroendocrinos del páncreas son lesiones raras se originan en las células de los islotes de Langerhans de dicha glándula. Mayoría son no funcionantes aunque un grupo importante pueden causar diversos síndromes clínicos dependientes de la hormona que secreten. Generalmente son malignos, su curso clínico es indolente. Pareciera que existe un aumento de su incidencia, lo que se encuentra asociado a una mejora de la tecnología diagnóstica por imágenes. El objetivo del trabajo es presentar el manejo clínico-quirúrgico de paciente femenina 41 años cuyo motivo de consulta fue ictericia de aparición brusca debida a lesión de ocupación de espacio a nivel de cabeza del páncreas por lo que se realiza pancreatoduodenectomía clásica sin complicaciones. Los estudios histológicos e inmunohistoquímicos denotan tumor neuroendocrino de bajo potencial de malignidad. En la actualidad el único tratamiento con intentos curativos para estas lesiones, continua siendo la cirugía radical con márgenes libres de enfermedad.
Palabras clave: Páncreas, tumores, neuroendocrinos, células insulares.
Abstract: The pancreatic neuroendocrine tumors are rare lesions that originate in the Langerhans islet’s cells of the gland. Most of them are nonfunctioning although a large group can cause various clinical syndromes dependent the hormone secreting. They are usually malignant although their clinical course is indolent. Currently it seems that there is an increase in the incidence, which is associated with an improvement in the diagnostic imaging technology. The aim of this work is to present the clinical and the surgical management of a female patient 41 years old, whose complaint was sudden onset jaundice due to occupying space lesion at the level of the head of the pancreas, so we realized the classical pancreatic-duodenectomy surgery without any complications. The histological and the immunohistochemically studies denote a neuroendocrine tumor of low malignant potential. Currently the only treatment with curative attempts to these tumors remains the radical surgery with disease free margins.
Keywords: Pancreas, neuroendocrine, tumors, islet cells.
INTRODUCCIÓN
Los tumores neuroendocrinos (TNE) de la glándula pancreática son lesiones raras que se originan en las células de los islotes de Langerhans (páncreas endocrino) constituyendo un grupo heterogéneo de tumores que producen hormonas activas por lo que se asocian a síndromes clínicos específicos. Aunque se han descrito más de 19 células activas desde el punto de vista endocrinológico capaces de producir hasta 50 aminas o péptidos diferentes; solo se conocen cinco síndromes clínicos bien definidos: insulinoma, gastrinoma, glucagonoma, vipoma y somatostatinoma(1) . En este sentido, la mayor parte de los TNE del páncreas son “no funcionantes” y se descubren de manera incidental durante los estudios por imágenes efectuados por otras razones.
Actualmente, la incidencia de los tumores neuroendocrinos pancreáticos, llamados así desde 2010, se considera bajo en comparación con otras entidades; constituyendo alrededor de 0,4 casos por cada 100 000 habitantes. Afecta a los hombres en una proporción ligeramente más alta, así como a los caucásicos, con un pico mayor de incidencia entre la sexta y la séptima década de la vida. Cuando se presentan en edades más tempranas están asociados con la neoplasia endocrina múltiple tipo 1 (MEN-1) o son lesiones funcionantes (2,3).
Inicialmente, estas lesiones pueden no dar síntomas clínicos y en aquellos casos donde los desarrollan son inespecíficos por lo que el diagnóstico siempre es tardío. Un número importante de estos tumores se malignizan, dan metástasis al hígado ocasionando la muerte del paciente. No obstante, cuando se comparan con su contraparte el adenocarcinoma pancreático, tienen un mejor pronóstico (3,4).
Su extrema rareza, heterogeneidad, comportamiento biológico errático y su naturaleza indolente hacen muy difícil poder implementar estudios aleatorizados acerca del cual sería el manejo clínico-quirúrgico más adecuado de estas lesiones, sobre todo y de manera muy especial en zonas como la nuestra donde el volumen de casos es mucho menor.
En tal sentido, es trascendente para el cirujano al encarar su manejo conocer de qué estamos hablando: ¿se trata de un TNE o de un adenocarcinoma?, ¿es funcionante o no funcionante?, ¿se localiza en la cabeza, cuerpo o cola del páncreas?, ¿qué tamaño tiene: es mayor o menor de 2 cm?, ¿qué características histológicas y bioquímicas presenta el tumor (angio-invasión, invasión a órganos vecinos, Ki 67)? Una vez identificado adecuadamente el tumor, el cirujano no solo debe estar técnicamente apto y capacitado para realizar resecciones complejas, sino que también debe ser intelectualmente competente en términos de manejar los conocimientos de los TNE para adoptar las mejores estrategias en este tipo particular de tumores (5).
El objetivo de la investigación radica en presentar el caso clínico de una paciente con un tumor neuroendocrino de la cabeza del páncreas (células insulares) no funcionante de baja malignidad, su abordaje quirúrgico, haciendo posteriormente un breve análisis de esta rara entidad nosológica
CASO CLÍNICO
Se trata de una paciente femenina de 41 años oriunda de la localidad, que inicia su enfermedad actual presentando tinte ictérico de piel y mucosas de aparición brusca por lo que recibe tratamiento médico inicial como hepatitis viral. En vista de la ausencia de mejoría consulta a gastroenterólogo quien le practica pancreato-colangiografía retrógrada endoscópica (ERCP) observando imagen de defecto de llenado a nivel del colédoco retro-pancreático (Figura 1). Durante dicho procedimiento le coloca una prótesis biliar para manejo del cuadro ictérico (Figura 2).

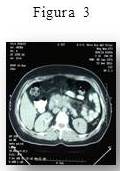
Es enviada a nuestra institución para su respectiva evaluación y adecuado tratamiento. Al interrogatorio niega antecedentes y otros datos de importancia. Durante la evaluación inicial se observa una paciente en buenas condiciones generales asintomática, sin ictericia al momento del examen, con todos los análisis de laboratorio dentro de la normalidad salvo el marcador tumoral CA 19-9 que se encontraba en 183,5 Ul/mL (Vn: 0-37 Ul/mL). Como extensión del examen físico se realiza ultrasonido abdominal en tiempo real, donde se distingue una lesión de ocupación de espacio (LOE) a nivel de la cabeza del páncreas de bordes romos, bien definidos, sin compromiso vascular ni otras imágenes sugestivas de metástasis locorregional; igual imagen se observa en la tomografía axial computarizada (Figura 3). Resto dentro de la normalidad.
Intervención: completados los exámenes preoperatorios y previo consentimiento informado se decide realizar pancreatoduodenectomía (Operación de Whipple) mediante laparotomía mediana. Hallazgos intraoperatorios: órganos intra-abdominales normales, se aborda el páncreas por la transcavidad de los epiplones encontrando en la cabeza del mismo, una lesión de aspecto tumoral de aproximadamente 8 cm x 6 cm de color blanco grisácea de consistencia dura, móvil no adherida a estructuras vasculares vecinas. En vista de estos hallazgos se procede a efectuar la pancreatoduodenectomía clásica que se completa sin complicaciones. Diagnóstico histopatológico: tumor de 3,5 cm x 2,5 cm mixto del páncreas: carcinoma de células insulares sólido asociado a adenocarcinoma ductal moderadamente diferenciado. Ganglios linfáticos y márgenes de resección sin evidencia de neoplasia. Inmunohistoquímica: neoplasia neuroendocrina pancreática bien diferenciada de bajo potencial de malignidad, Ki-67: 14 % ( de células insulares ).
La paciente evoluciona favorablemente encontrándose hasta la actualidad sin evidencia de enfermedad. Con base a los resultados de inmunohistoquímica se efectuaron análisis de gastrina, insulina y somatostatina los cuales resultaron dentro de la normalidad. No recibió tratamiento adicional.
DISCUSIÓN
Los tumores neuroendocrinos del páncreas son lesiones raras que teóricamente se originan de células pluri-potenciales ubicadas tanto en los islotes de Langerhans como en el sistema ductal de dicha glándula (6), con una incidencia de 1-1,5 por cada 100 000 habitantes. Cerca del 50 % de estos tumores secretan uno o más péptidos activos desde el punto de vista biológico, originando diversos síntomas clínicos sistémicos según la sustancia bioquímica secretada (llamados tumores neuroendocrinos o de células insulares funcionales). A causa de lo inespecífico de los síntomas, el diagnóstico suele generalmente retardarse. De igual forma, en el caso de los tumores no funcionantes el diagnóstico se retarda mucho más y cuando se diagnostican se hace con base a los síntomas causados por el crecimiento y volumen tumoral como el presente caso (7).
Se han descritos diversos tipos de tumores de células insulares funcionantes. Según las características bioquímicas de los péptidos liberado por ellos los más conocidos son: insulinoma que secretan pro-insulina lo que ocasiona síntomas relacionados con hipoglicemia; gastrinoma secretores de gastrina y conexo con enfermedad ulcerosa péptica; glucagonoma productores de glucagón y asociados a diabetes, dermatitis, trombosis venosa profunda y depresión; vipoma productor de péptido intestinal vasoactivo originando el síndrome de diarrea acuosa, hipopotasemia y aclorhidria también conocido como síndrome de Verner-Morrison y el somatostatinoma extremadamente raro asociado a diabetes, litiasis vesicular, pérdida de peso, diarrea y esteatorrea (1,8) .
Su contraparte los tumores de células insulares no funcionantes como el presente caso de estudio, en su mayoría son malignos (90 %), aunque también secretan sustancias tales como, cromogranina A, enolasa neuro-específica, grelina ente otros, no presentan los síndromes clínicos como los anteriormente descritos. La generalidad de estos pacientes se diagnostican incidentalmente durante una evaluación mediante estudios por imagen; cuando aumentan de tamaño los síntomas más frecuentes son: dolor abdominal (35 %-78 %), disminución peso (20 %-35 %), anorexia y náuseas (45 %), ictericia obstructiva (17 %-40 %), hemorragia intra-abdominal (4 %-20 %), masa abdominal palpable (7 %-40 %) (8).
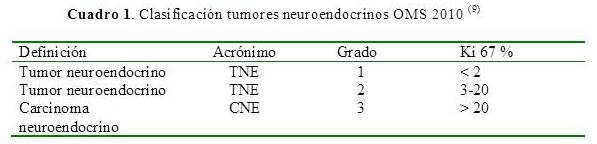
Recientemente, la Organización Mundial de la Salud (OMS) ha clasificado los TNE del páncreas de acuerdo a su grado de diferenciación en bien diferenciados que comprende a los tumores neuroendocrinos de comportamiento benigno y los mal diferenciados que incluye a los carcinomas neuroendocrinos de peor pronóstico. En esta nueva clasificación se introduce el concepto del comportamiento biológico del tumor con base al índice de proliferación celular o índice mitótico Ki-67 como medida de la velocidad de crecimiento tumoral (2). A mayor índice mitótico peor pronóstico (Cuadro 1). Tumores con Ki67 menores de 20 % se consideran benignos y con buen pronóstico; lesiones con Ki 67 mayores a 20 % son malignas y su pronóstico es peor.
A pesar de una alta tasa de metástasis hepáticas al momento del diagnóstico, los TNE pancreáticos no funcionantes se asocian con un pronóstico más favorable en comparación con muchos otros tumores malignos gastrointestinales. En general, la supervivencia a los 5 años varía del 27 % al 33 % en grandes series (10,11).
En este sentido, se han reportado casos donde el manejo agresivo está asociado con resultados más favorables, incluso en pacientes con metástasis hepáticas. Así lo demostraron Franko y cols., en una reciente revisión tomada de la base de datos del Instituto Nacional de Cáncer (Surveillance, Epidemiology and end Results Program: SEER) analizando más de dos mil pacientes con tumores TNE pancreáticos no funcionantes donde observaron una mediana de supervivencia de 4,8 años en pacientes con enfermedad metastásica sometidos a tratamiento quirúrgico agresivo, en comparación con 1 año de los pacientes que no se sometieron a la resección (10).
En la actualidad, el único tratamiento curativo de los TNE pancreáticos o de células insulares no funcionales es la resección quirúrgica completa. Se ha aconsejado retirar la mayor cantidad de tumor posible en caso de enfermedad no resecable como medida paliativa, observando supervivencias a cinco años en pacientes sometidos a resecciones incompletas (12,13). Como se mencionó anteriormente, el tratamiento de elección tanto para la enfermedad localizada como para las lesiones localmente avanzadas la piedra angular es la cirugía cuando es técnicamente posible (4,5,14). Por lo general, el enfoque quirúrgico de los TNE del páncreas depende de la ubicación dentro de la glándula y el tamaño de la lesión. Las complicaciones posoperatorias suelen ser frecuentes y agravadas por la secundaria insuficiencia endocrina pancreática (14). Por lo general, el enfoque quirúrgico de los TNE del páncreas depende de la ubicación dentro de la glándula y el tamaño de la lesión. Las complicaciones posoperatorias suelen ser frecuentes y agravadas por la secundaria insuficiencia endocrina pancreática (14).
Teniendo en cuenta los avances de las técnicas de diagnósticas por imágenes, se ha producido una mejora significativa para la localización de las lesiones ≤ 2 cm, de las cuales aproximadamente el 5 % son malignas. La extirpación quirúrgica de estas lesiones pequeñas podría lograrse mediante resecciones no convencionales como las enucleaciones, con un menor riesgo de disfunción endo / exocrina (15,16,17). Aunque este procedimiento no permitiría una correcta evaluación de la afectación ganglionar, sin embargo, ha mostrado datos similares de supervivencia a los 5 y 10 años, en comparación con las resecciones pancreáticas tradicionales(18,19). Durante la realización de la enucleación y/o cualquier otro procedimiento conservador, el riesgo de fístulas pancreáticas puede ser mayor que con otros enfoques, sin embargo, la gravedad de estas complicaciones es por lo general inferior a la observada en los procedimientos quirúrgicos clásicos (13,20).
En el contexto de lesiones localmente avanzadas, la cirugía puede ser considerada en casos sin afectación de la vena porta (cavernomatosis) o de los vasos mesentéricos superiores. Este enfoque radical y agresivo es apoyada por estudios previos que han demostrado un claro beneficio en la supervivencia en casos de resecciones extensas donde la enfermedad residual macroscópica se puede evitar (R2) (10,13,21).
Finalmente, en el caso singular de la neoplasia endocrina múltiple tipo 1 conocida por las siglas MEN-1(Hiperplasia paratiroidea, adenoma hipofisario y tumor neuroendocrino pancreático), donde la mayoría de las lesiones del páncreas son de tipo no-funcionante, existe un consenso general de que el abordaje quirúrgico debe ser como el señalado anteriormente sobre todo si las lesiones son mayores de 2 cm, sintomáticas, con riesgo de progresión a distancia, dolorosas o con un crecimiento anual mayor de 0,5 cm. Por otra parte, cuando las lesiones pancreáticas son menores a 2 cm, se recomienda la vigilancia mediante ultrasonido endoscópico con base al alto porcentaje de multifocalidad que presentan estas lesiones cuando se encuentran asociadas al síndrome hereditario MEN-1 (22,23,24,25).
Referencias
1. Vargas C, Llano R. Tumores neuroendocrinos gastroenteropancreáticos. Rev Col Gastroenterol. 2010;25(2):165-176.
2. Viudez A, Acosta A, Carvalho F, Vera R, Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:93-206.
3. Burgos L, Burgos M. Tumores neuroendocrinos del páncreas. Rev Méd Chile. 2004;132:627 634.
4. Meadro R, Alvarado I, González D, López S, Páez F. Tumor neuroendocrino gastro-entero-pancreático. Factores pronósticos quirúrgicos. Rev Méd Inst Mex Seguro Soc. 2012;50(3):243-248.
5. Huertas E, Sánchez F. Carcinoma neuroendocrino de páncreas. ¿Debe ser abordado igual que un adenocarcinoma? Acta Gastroenterol Latinoam. 2009;39(1):39-41.
6. Vortmeyer A, Huangs S, Lubensky I, Zhuang Z. Non-islet origin of pancreatic islet cell tumors. J Clin Endocrinol Metab. 2004;89(4):1934-1938.
7. Brentjens R, Saltz L. Islet cell tumors of the pancreas: The medical oncologist's perspective. Clin Chirurg North Am. 2001;81(3):527-542.
8. Grenzinsky S, Mazeh D, Gross D. Clinical features of pancreatic neuroendocrine tumors. J Hepatobiliary Pancreat Sci. 2015;22:578-585.
9. Bosman FT, Carneiro F, Hruban RH, Theise ND, editores. WHO Classification of tumor of the digestive system. 4a edición. Francia:Lyon IARC Press;2012.
10. Franco J, Feng W, Yip L, Genovese E, Moser A. Non-functional neuroendocrine carcinoma of the pancreas: Incidence, tumor biology, and outcomes in 2 158 patients. J Gastrointest Surg. 2010;14(3):541-548.
11. Fraenkel M, Kim M, Faggiano A, Valk G. Epidemiology of gastroenteropancreatic neuroendocrine tumors. Best Pract Res Clin Gastroenterol. 2012;26(6):691-703.
12. Barno P, Satre M, Ballestero A, Molina J, Lisa E, Medía E, et al. Tumores neuroendocrinos pancreáticos: Manejo multidisciplinario y revisión de la literatura. Rev Arcial. 2015;2(2):39-54.
13. Folkert I, Hernandez P, Roses R. Multidisciplinary management of nonfunctional neuroendocrine tumor of the pancreas. World J Gastroenterol. 2016;22(11):3105-3116.
14. Hill J, McPhee J, McDade T, Zhou Z, Sullivan M, Whalen G, et al. Pancreatic neuroendocrine tumors: The impact of surgical resection on survival. Cancer. 2009;115(4):741-751.
15. Smith J, Ng S, Hill J, Simons J, Arous E, Shah S, et al. Complications after pancreatectomy for neuroendocrine tumors: A national study. J Surg Res. 2010;163(1):63-68.
16. Bettini R, Partelli S, Boninsegna L, Capelli P, Crippa S, Pederzoli P, et al. Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery. 2011;150(1): 75-82.
17. Falconi M, Matovani W, Cirppa S, Mascetta G, Salvia R, Pederzoli P. Pancreatic insufficiency after different resections for benign tumors. Br J Surg. 2008;95(1):85-91.
18. Cauley C, Pitt H, Ziegler K, Nakeeb A, Schmidt C, Zyromski N, et al. Pancreatic enucleation: Improved outcomes compared to resection. J Gastrointest Surg. 2012;16(7):1347-1353.
19. Casadei R, Ricci C, Rega D, D´Ambra M, Pezilli R, Tomassetti P, et al. Pancreatic endocrine tumors less than 4 cm in diameter: Reset or enucleate? A single center experience. Pancreas. 2010;39(6):825-828.
20. Cherif R, Gaujoux S, Couverlard A, Dokmak S, Vuillerme M, Ruszniewski P, et al. Parenchyma-sparing resections for pancreatic neuroendocrine tumors. J Gastrointest Surg. 2012; 16(11):2045-2055.
21. Galindo R, Gabrielli N, Barros C, Moisan P, Martínez M, Torres M, et al. Tumores neuroendocrinos del páncreas: Resultados quirúrgicos y sobrevida alejada. Rev Chil Cir. 2013; 65(3):228-235.
22. Gauger P, Scheiman J, Wamsteker E, Richards M, Doherty G, Thompson N. Role of endoscopic ultrasonography in screening and treatment of pancreatic endocrine tumors in asymptomatic patients with multiple endocrine neoplasia type 1. Br J Surg. 2003;90(6):748-754.
23. Jensen R, Berna M, Bingham D, Norton J. Inherited pancreatic endocrine tumor syndrome: Advances in molecular pathogenesis, diagnosis, management and controversies. Cancer. 2008; 113(Supl 7):S1807-1843.
24. Batcher E, Madaj P, Gianoukakis A. Pancreatic neuroendocrine tumors. Endocr Res. 2011;36(1):35-43.
25. Amin S, Kim M. Islet cell tumors of the pancreas. Gastroenterol Clin North Am. 2016;45:83-100.
Notas de autor
wilmardejesus@gmail.com