Tumor maligno de la vaina del nervio periférico primario de mama informe de caso Unidad de Mastología Clínica Leopoldo Aguerrevere
Tumor maligno de la vaina del nervio periférico primario de mama informe de caso Unidad de Mastología Clínica Leopoldo Aguerrevere
Revista Venezolana de Oncología, vol. 29, núm. 2, pp. 123-129, 2017
Sociedad Venezolana de Oncología
Recepción: 10 Octubre 2016
Revisado: 12 Noviembre 2016
Aprobación: 12 Enero 2017
Resumen: Los tumores malignos de la vaina del nervio periférico representan entre 5 %-10 % del total de tumores malignos de tejidos blandos. Frecuentemente se asocian a neurofibromatosis tipo I aparecen principalmente en extremidades y tronco. En la mama, los sarcomas primarios constituyen 1 % del total de tumores malignos. CASO CLÍNICO: Paciente femenina de 30 años de edad con tumoración de 3 meses de evolución en mama derecha, de crecimiento rápido, sin antecedentes de neurofibromatosis. El diagnóstico en biopsia por punción fue: “hallazgos morfológicos sugestivos de lesión maligna con rasgos sarcomatosos”. A la paciente se le practicó mastectomía total simple con reconstrucción del colgado miocutáneo del músculo dorsal ancho, colocación de prótesis y simetrización de mama contralateral. El diagnóstico histopatológico final basado en estudio de inmunohistoquímica fue: “tumor maligno de la vaina del nervio periférico CONCLUSIÓN: Son escasos los casos reportados en la literatura de primario de la mama y aún más, en ausencia de neurofibromatosis tipo 1, lo que hace especial nuestro caso. El diagnóstico de estas lesiones debe hacerse sobre la base de un amplio muestreo en el espécimen de resección completa y con el apoyo de la inmunohistoquímica.
Palabras clave: Sarcoma, mama, tumor maligno, vaina del nervio periférico, neurofibromatosis.
Abstract: The malignant peripheral nerve sheath tumor represents 5 % - 10 % of all soft tissue sarcomas. They are commonly arising in association with the neurofibromatosis type I. The most common sites of origin include the extremities and the trunk. Breast primary sarcomas constitute 1 % of all mammary malignancies. CLINICAL CASE: A 30 year old female presented with a rapidly growing tumor in the right breast with 3 months of evolution. The histopathologic diagnostic for the ultrasound guided and core needle biopsy was: Morphological findings suggestive of malignant lesion with sarcomatoide features. A total mastectomy was performed with immediate reconstruction with a latissimus dorsi flap and simetrizacion of the contralateral breast and install prosthesis. The final histopathologic diagnostic based on the immunohistochemically study concluded: Malignant peripheral nerve sheath tumor. CONCLUSION: There are few cases reported in the literature of the malignant peripheral nerve sheath tumor localized primary of the breast, and even more, in the absence of the neurofibromatosis type 1, what makes our clinical case special. The diagnosis of these lesions should be based on extensive sampling of the complete resection specimen and they can be supported by immunohistochemistry.
Keywords: Sarcoma, breast, malignant tumor, peripheral nerve sheath, neurofibromatosis.
INTRODUCCIÓN
Los tumores de la vaina del nervio periférico son lesiones que se derivan de las células de Schwann o de las células pluripotenciales de la cresta neural (1). Estos tumores corresponden un espectro de entidades clínico patológicas bien definidas que comprenden tanto tumores benignos como el Schwannoma, hasta lesiones de elevado potencial maligno como el tumor maligno de la vaina del nervio periférico (TMVNP) (2).
Antiguamente, según la clasificación de la WHO 2007 de los tumores del sistema nervioso, los TMVNP podían clasificarse en grado II, III o IV. Sin embargo, actualmente son incluidos dentro del capítulo de tumores de tejidos blandos en el que no se establece esa diferencia sino que se clasifican como tumores de bajo y alto grado. Ellos representan sólo el 5 %-10 % del total de tumores malignos de los tejidos blandos (2,3,4). Entre un 50 %-75 % de estos tumores aparecen a partir de la transformación de un neurofibroma, frecuentemente con características plexiformes o en el contexto de una neurofibromatosis tipo I, principalmente en cabeza y cuello (1,2,5). Los TMVNP que aparecen de novo usualmente se observan en el tronco o las caderas, comprometiendo el nervio ciático (2). También se ha descrito la aparición de TMVNP asociado a tratamiento radiante (6).
En la mama, los tumores que principalmente la afectan tienen su origen en el componente epitelioide-glandular de la misma. Los sarcomas primarios de la mama son raros y constituyen no más del 1 % de todos los tumores malignos de la mama (7). Dado lo infrecuente de estas lesiones, específicamente de esta entidad en la mama, nos hemos propuesto reportar nuestro caso.
CASO CLÍNICO
Paciente femenina de 30 años de edad presenta una masa tumoral en mama derecha de crecimiento rápido de 3 meses de evolución, no tiene antecedentes de enfermedad de neurofibromatosis en el grupo familiar ni ella lo presenta. Al examen físico se encuentra una masa tumoral de 16 cm. En diámetro mayor, firme no adherida a la piel ubicada en unión de cuadrantes externos (Figura 1). La paciente venía con una mamografía (Figura 2) y una ecografía mamaria (Figura 3)
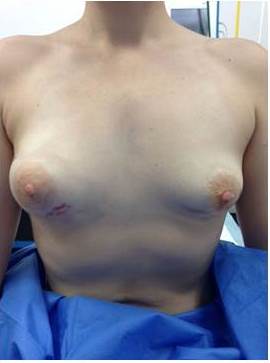
Figura 1
Mama derecha con masa tumoral en unión de cuadrante ínfero externo
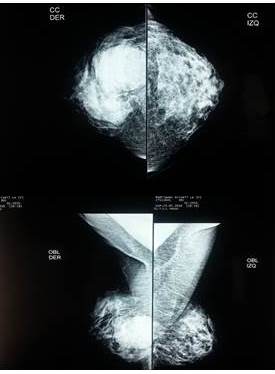
Figura 2
Mamografía fibroglandular densa en la que se observa una masa redondeada visible con bordes irregulares en cuadrante ínfero externo.
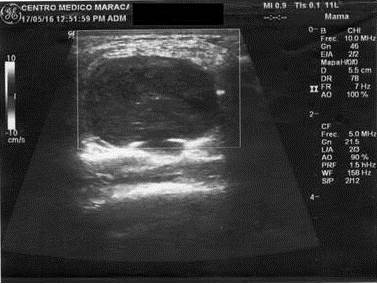
Figura 3.
Mama con tejido fibro-ductal con distribución difusa, imagen nodular dominante de aspecto sólido en mama derecha hipoecogénica ovalada con fino septo lineal a nivel de cuadrantes ínfero externo
Se realizó biopsia por punción con aguja de corte (trucut 14G) guiada por ultrasonido, en la que se obtuvieron múltiples fragmentos de la lesión. El mayor de 1 cm de longitud por 0,2 cm de diámetro, los cuales fueron fijados en formol buffer al 10 % y procesados en nuestro laboratorio de patología. Se realizaron cortes histológicos seriados de 3µ con coloración de hematoxilina-eosina y se emitió una primera impresión diagnóstica: “hallazgos morfológicos sugestivos de lesión maligna con rasgos sarcomatosos”, en la que se describió como una lesión constituida por células de aspecto neoplásico con núcleos pequeños, citoplasma claro y bordes citoplasmáticos indistintos, con actividad mitótica de 8 mitosis en 10 campos de mayor aumento. Dado el tamaño de la lesión y su evolución, se sugirió realizar extirpación de la misma para su completo estudio histopatológico y realización de estudio de inmunohistoquímica a fin de poder emitir un diagnóstico concluyente.
A la paciente se le realizó una mastectomía total simple con reconstrucción de un colgajo miocutáneo del musculo dorsal ancho, colocación de prótesis y simetrización de la mama contralateral. (Figura 4)
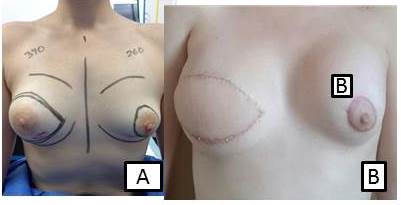
Figura 4
A: Marcaje de la paciente para practicar la mastectomía total simple y planificación de la reconstrucción B: Resultado del posoperatorio inmediato.
La pieza quirúrgica de la mastectomía total fue procesada en el laboratorio de la unidad de patología, observándose en el estudio macroscópico (Figura 5) hacia unión de cuadrantes externos y a 0,2 cm del borde de resección profundo (músculo), una lesión de aspecto tumoral de 16,5 cm x 6 cm x 5 cm, de bordes bien delimitados, pardo-amarillenta, con áreas de hemorragia, siendo el resto del tejido mamario de aspecto usual. El diagnóstico histopatológico fue: “sarcoma de alto grado (grado 2), según la clasificación de la Federación Francesa del Centro de Cáncer Grupo de Sarcomas”, con actividad mitótica de 20 mitosis en 10 campos de mayor aumento, presencia de necrosis en menos del 50 %, con áreas densamente celulares de células fusiformes alternas con otras menos celulares y presencia de células epitelioides (Figura 5). Los bordes de resección de la mastectomía estuvieron libres de lesión.
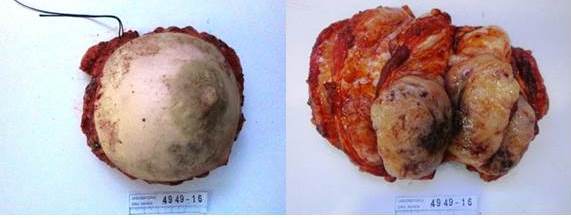
Figura 5
Pieza de mastectomía total simple, Tamaño tumoral: 16,3 cm. Bordes libres de tumor.
Se realizó estudio de inmunohistoquímica con polímero marcado con peroxidasa conjugada al anticuerpo secundario, realizándose inmunorreacciones para los siguientes anticuerpos: vimentina, citoqueratina AE1/AE3 (CKAE1/AE3), actina músculo liso (AML), desmina, CD34, proteína S100, enolasa, receptores de estrógenos (RE), receptores de progesterona (RP) y Ki67. Utilizando controles previamente conocidos como positivos y controles internos, observándose en las células de la lesión: vimentina positiva. CD34 positivo multifocal. Proteína S100 positiva focal (Figura 6). Siendo la CKAE1/AE3, AML y desmina negativos en las células tumorales. El Ki67 se demostró una actividad proliferativa del tumor del 70 %. Por lo que el caso se concluyó como: “hallazgos morfológicos e inmunohistoquímicos compatibles con TMVNP
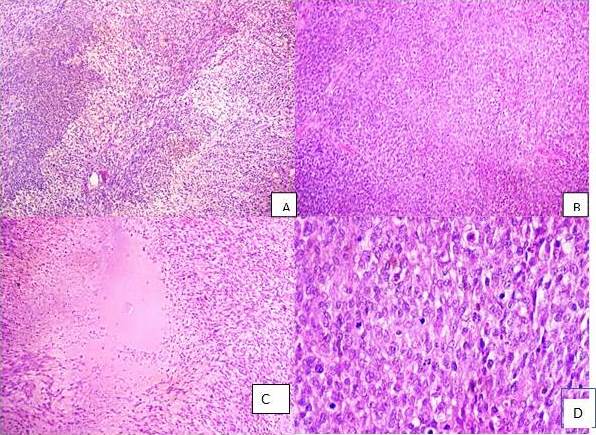
Figura 6
Microfotografías de la lesión en hematoxilina-eosina A: Presencia a áreas más celulares alternando con áreas menos celulares. Aumento 100 x. B: Áreas densamente celulares con presencia de células de aspecto epitelioide. Aumento 100 x. C: Áreas de necrosis con empalizada de núcleos neoplásicos. Aumento D: Elevada actividad mitótica. Aumento 400
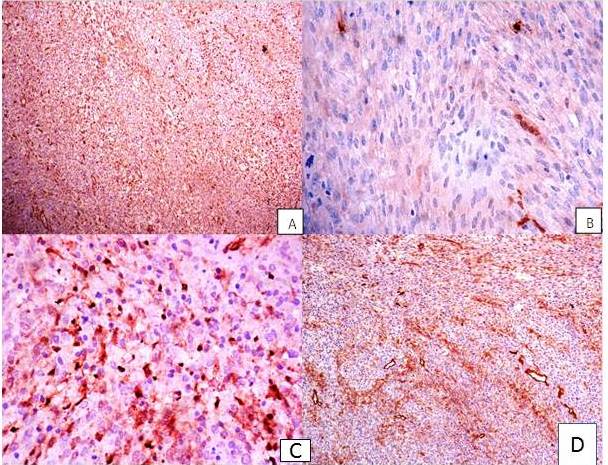
Figura 7
Microfotografías de las diferentes inmunorreacciones A: VIMENTINA, con marcaje citoplasmático difuso fuerte de las células neoplásicas. Aumento 100x B: PROTEÍNA S100 con positividad citoplasmática y nuclear en aisladas células neoplásicas. Aumento 400x C: ENOLASA positividad citoplasmática granular en algunas células neoplásicas. Aumento 400x. D: CD34 positividad multifocal de membrana citoplasmática en las células neoplásicas. Aumento 100x.
DISCUSIÓN
El término TMVNP actualmente reúne a aquellos tumores que se originan de las células que constituyen las vainas de los nervios, tales como las células de Schwann o las células perineurales y sustituye términos antiguamente utilizados como: Schwannoma maligno, neurilemmoma maligno y neurofibrosarcoma. Ellos representan entre el 5 %-10 % de todos los sarcomas de los tejidos blandos (2,3.4). Su incidencia en la población general es del 0,001 %, la cual puede verse incrementada hasta en un 42 % en aquellos pacientes con neurofibromatosis tipo 1, con mayor tendencia a la malignización de los neurofibromas de localización profunda (8,9). La etiología de cerca del 11 % de los TMVNP se asocia al tratamiento radiante, tras la irradiación de los nervios periféricos por otra causa con un periodo de latencia posterior a la irradiación de entre 10 y 20 años. La edad de presentación está descrita entre los 20 a 50 años de edad y los sitios de origen más frecuentes incluyen las extremidades y el tronco, principalmente comprometiendo el nervio ciático, el plexo braquial y el plexo sacro, con un tamaño promedio que generalmente supera los 5 cm (10). La tasa de supervivencia descrita para estos tumores suele ser muy pobre a los 5 años (11).
El TMVNP de la mama es una lesión extremadamente rara, con muy pocos casos publicados en la literatura. El diagnóstico definitivo de esta entidad solo puede hacerse sobre la base de una adecuada interpretación de la inmunohistoquímica que se correlacione con los hallazgos morfológicos y permita hacer los diagnósticos diferenciales, entre ellos: El tumor filodes maligno, fibrosarcoma y leiomiosarcoma (8). Sin embargo, el tumor filodes maligno es una entidad bifásica con un componente estromal maligno y un componente epitelial benigno. El tumor filodes maligno exhibe los típicos espacios con forma de “hoja de helecho” revestidos por un epitelio ductal benigno, en ocasiones comprimido y colapsado por el sobre-crecimiento estromal. A diferencia de estas lesiones, el TMVNP puede exhibir ocasionales ductos mamarios que constituyen ductos “atrapados” dentro de la lesión, sin la conformación característica del componente epitelial que exhibe el tumor filodes (12).
Establecer el diagnóstico diferencial entre el TMVNP, el fibrosarcoma y leiomiosarcoma puede ser aún más retador. Porque estas lesiones están típicamente constituidas por una población fusocelular principalmente. A pesar que académicamente los tumores de origen neural se distinguen por la característica “ondulante” de su célula y el leiomiosarcoma por tener citoplasma más eosinofílico con núcleos menos aguzados respecto al fibrosarcoma, estas entidades poseen diferentes variantes, entre ellas la presencia de células “epitelioides” que pueden oscurecer el diagnóstico morfológico.
Por otra parte, establecer el diagnóstico diferencial entre un tumor benigno de la vaina del nervio periférico (neurofibroma) y su contraparte maligna (TMVNP), puede ser complejo, ya que algunos neurofibromas pueden ser bastante celulares e incluso tener ocasionales células pleomórficas. Sin embargo, el contaje mitótico en estos casos resulta fundamental para establecer el diagnóstico ya que los TMVNP exhiben distintivamente elevada actividad mitótica (3,4,5).
Para establecer de manera adecuada los diagnósticos diferenciales en estos casos es de relevante obligatoriedad realizar un amplio y exhaustivo muestreo de estas lesiones, por lo que realizar este tipo de diagnóstico en material de biopsia percutánea o incisional es limitado e impreciso, aumentando de forma importante la posibilidad de diagnóstico errado.
Desde el punto de vista inmunohistoquímico, la proteína S100 es tradicionalmente usada como el marcador más sensible para determinar el origen neural de las lesiones. Sin embargo, en los TMVNP tiene una utilidad relativamente limitada, descrita en un amplio rango entre el 50 %-90 % (13), dependiendo del grado de diferenciación del tumor. Los TMVNP de alto grado pueden exhibir solo escasa y en algunos casos ninguna inmunorreactividad a este marcador (2,14). La presencia de células epitelioides en algunos TMVNP puede condicionar una positividad en esas áreas del marcador epitelial CKAE1/AE3. El marcador mesenquimático vimentina suele ser típicamente positivo en el citoplasma de las células fusiformes (15,16). Focalmente además, algunos TMVNP así como algunos neurofibromas pueden mostrar grupos celulares inmunorreactivos al CD34 indicando la heterogeneidad celular de estos tumores (18) y la similaridad sugerida por algunos autores entre los neurofibromas y los TMVNP (17,18).
La negatividad a la desmina y a la AML descartó el origen muscular de nuestro tumor en estudio. A pesar de la posibilidad de positividad descrita al CD34 para algunos los tumores filodes (19,20), la ausencia del patrón morfológico y la positividad focal a la proteína S100 nos permitió descartar esta posibilidad diagnóstica. Por otra parte, el diagnóstico de fibrosarcoma constituye un diagnóstico de exclusión, que no aplicó en este caso.
El tratamiento descrito para estas lesiones a pesar de la poca experiencia con ellos por lo infrecuente de su presentación es la mastectomía total seguida de radioterapia. No se incluye dentro del protocolo la disección axilar dado que la vía de diseminación de estas lesiones es hematológico y no linfática (11).
El TMVNP es una entidad de muy rara presentación en la mama, con muy escasos casos reportados en la literatura en dicha localización. La mayoría de los TMVNP son descritos en pacientes con neurofibromatosis tipo 1, diagnóstico que nuestra paciente no comparte. Este por tanto, constituye uno de los muy escasos casos reportados en la literatura de TMVNP de la mama en una paciente sin diagnóstico de neurofibromatosis tipo 1 asociado.
Referencias
1. Rodríguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319.
2. Lois DN, Ohgaki H, Wiestler O, Cavenee W, Burger P, Jouvet A, et al. WHO Classification of tumors pathology and genetics of tumors of the nervous system. Acta Neuropathol. 2007;114(2):97-109.
3. Weiss SW, Goldblum JR. Malignant tumors of the peripheral nerves. En: Strauss M, Grey L, editores. Enzinger and Weiss's Soft Tissue Tumors 4a edition. St. Louis: Mosby, Inc; 2001.p.1209-1264.
4. Woo OH, Yong HS, Lee JB, Kim A, Koo BH, Kang EY. A giant malignant peripheral nerve sheath tumor of the breast: CT and pathological findings. Br J Radiol. 2007;80(950):e44-e47.
5. Dhingra KK, Mandal S, Roy S, Khurana N. Malignant peripheral nerve sheath tumor of the breast: Case report. World J Surg Oncol. 2007;21(5):142.
6. Baehring JM, Betensky RA, Batchelor TT. Malignant peripheral nerve sheath tumor: The clinical spectrum an outcome of treatment. Neurology. 2003;61(5):696-698.
7. Rosen PP, editor. Sarcoma. Filadelfia: Lippincott Williams and Wilkins; 2001.
8. Thanapaisal C, Koonmee S, Siritunyaporn S. Malignant peripheral nerve sheath tumor of the breast in a patient without neurofibromatosis: A case report. J Med Assoc Thai. 2006;89(3):377-379.
9. Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573-1577.
10. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinic pathologic study of 120 cases. Cancer. 1986;57:2006-2021.
11. Yang JC, Baker AR, Sindelar WF, Danforth DN, Topalian SL, DeLaney T, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol. 1998;16:197-203.
12. Yandan L, Jia Z, Jianlun L, Huawei L, Yi J, Wei W. Periductal stromal sarcoma of the breast: A case report and review of the literature. Oncol Lett. 2014;8(3):1181-1183.
13. Stasik CJ, Tawfik O. Malignant peripheral nerve sheath tumor with rhabdomyosarcomatous differentiation (malignant triton tumor). Arch Pathol Lab Med. 2006;130:1878-1881.
14. Ramanathan RC, Thomas JM. Malignant peripheral nerve sheath tumors associated with von Recklinghausen's neurofibromatosis. Eur J Surg Oncol. 1999;25:190-193.
15. Guo A, Liu A, Wei L, Song X. Malignant peripheral nerve sheath tumors: Differentiation patterns and immunohistochemically features - A mini review and our new findings. J Cancer. 2012;3:303-309.
16. Hirose T, Tani T, Shimada T, Ishizawa K, Shimada S, Sano T. Immunohistochemically demonstration of EMA/Glut1-Positive perineural cells and CD34-Positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16(4):293-298.
17. Herrera GA, de Moraes HP. Neurogenic sarcomas in patients with neurofibromatosis (von Recklinghausen’s disease). Light, electron microscopy and immunohistochemically study. Virchows Arch A Pathol Anat Histopathol. 1984;403:361-376.
18. Hirose T, Hasegawa T, Kudo E, Seki K, Sano T, Hizawa K. Malignant peripheral nerve sheath tumors: An immunohistochemically study in relation to ultrastructural features. Hum Pathol. 1992;23:865-870.
19. Aranda F, Laforga JB, Lopez JI. Phyllodes tumor of the breast. A immunohistochemically study of 28 cases with special attention to the role of myofibroblast. Pathol Res Pract. 1994;190:474-481.
20. Auger M, Hanna W, Kahn HJ. Cystosarcoma phyllodes of the breast and its mimics. An immunohistochemically and ultrastructural study. Arch Pathol Lab Med. 1989;113:1231-1235.
Notas de autor
drgerher@gmail.com