SARCOMA DE EWING: EXPERIENCIA EN 25 PACIENTES TRATADOS DE MANERA MULTIDISCIPLINARIA
SARCOMA DE EWING: EXPERIENCIA EN 25 PACIENTES TRATADOS DE MANERA MULTIDISCIPLINARIA
Revista Venezolana de Oncología, vol. 30, núm. 2, pp. 98-108, 2018
Sociedad Venezolana de Oncología

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 4.0 Internacional.
Recepción: 15 Enero 2018
Revisado: 02 Febrero 2018
Aprobación: 03 Marzo 2018
Resumen: OBJETIVO: Presentar la experiencia en 25 pacientes menores de 18 años con diagnóstico de sarcoma de Ewing quienes recibieron tratamiento multidisciplinario. MÉTODO: Revisión retrospectiva de 25 pacientes tratados con cirugía, quimioterapia y radioterapia en período 2000-2015. 25 pacientes recibieron quimioterapia. 24 recibieron radioterapia. 1 con lesión localizado fue resecado completamente y sólo recibió quimioterapia. El esquema de quimioterapia más usado fue: vincristina, doxorrubicina, ciclofosfamida con ifosfamida, etopósido. Recibieron RTC3D 14, RT2D 6, RTIM 4. La dosis empleada fue: 4 140 cGy en enfermedad microscópica y 5 040 cGy en enfermedad macroscópica. RESULTADOS: De 25 pacientes: 18 tuvieron enfermedad localizada (68,1 %): 8 óseo, 7 extraóseo, 3 tumor de Askin. 9 VSE, 2 VCE, 6 MCE, 1 MSE. 7 con enfermedad metastásica, todos de localización ósea, sólo 1 VCE. La SG a los 5 años 36,8 % y SLE 34,4 %. La SG a 5 y 10 años para enfermedad localizada 75,6 % y 50,4 % y SLE a 5 años 48,7 %. La SG en enfermedad metastásica a 2 años 28,6 % y SLE 14,3 %. 1 paciente con tumor localizado falleció por LMA después de quimioterapia sin SE, 1 presentó condrosarcoma, 15 años posterior a tratamiento con quimioterapia, radioterapia y 1 presentó escoliosis severa. CONCLUSIONES: El tratamiento con cirugía, quimioterapia y radioterapia representa el estándar de tratamiento para pacientes con diagnóstico de sarcoma de Ewing. Los resultados de la presente serie se comparan con los reportados por otros investigadores.
Palabras clave: Cáncer, sarcoma, tratamiento, radioterapia, quimioterapia, cirugía, sobrevida.
Abstract: OBJECTIVE: Report the experience in 25 patients less than 18 years with diagnosis Ewing sarcoma who received multidisciplinary treatment. METHOD: Retrospective review of 25 patients treated with surgery, chemotherapy, radiotherapy in 2000-2015. 25 patients received chemotherapy. 24 received radiotherapy. 1 with localized tumor had wide local resection only received chemotherapy. Chemotherapy regimen most frequently employed was vincristine, doxorrubicin, cyclophosphamide, and ifosfamide, etoposide. Radiotherapy was given using 3DCRT in 14, 2DRT in 6, and IMRT 4. The dose employed was 4 140 cGy in microscopic disease and 5 040 cGy in macroscopic disease. RESULTS: 25 patients, 18 had localized disease (68.1 %): osseous 8, extra-osseous 7 and Askin tumor 3, of these, 9 are alive without tumor, 2 alive with lesion, 6 dead with sarcoma, 1 dead without tumor. 7/25 had metastatic disease , all located in osseous sites, only one is alive without lesion. The 5 years OS was 36.8 % and EFS 34.4 %. The 5-10 years OS was 75.6 % and 50.4 % for localized disease, the 5 years EFS 48.7 %. For metastatic disease the 2 years OS was 28.6 % and EFS 14.3 %. One patient with ES died with AML after chemotherapy without ES, one patient developed chondrosarcoma fifteen years after radiotherapy and chemotherapy, and one patient scoliosis. CONCLUSIONS: Surgery, chemotherapy and radiotherapy represent the treatment standard in ES. The results reported in the present series are compared with the results of other international institutions.
Keywords: Cancer, sarcoma, treatment, radiation therapy, chemotherapy, surgery, survival.
INTRODUCCIÓN
El sarcoma de Ewing (SE), es el tumor primario del hueso más común después del osteosarcoma, representando alrededor del 3 % de las neoplasias malignas pediátricas. Aproximadamente, 40 % de los casos ocurren durante la pubertad, 30 % antes de los 10 años y 5 % después de los 20 años. Existe una discreta preponderancia de la enfermedad en varones. El SE no ha sido asociado de manera consistente con síndromes genéticos familiares o congénitos (1).
El sarcoma de Ewing fue descrito por James Ewing en 1921 famoso patólogo del Centro de Cáncer Memorial Sloan-Kettering en Nueva York, como tumor indiferenciado que involucra la diáfisis de hueso largos, tumor de origen óseo compuesto de capas de células pequeñas redondas muy sensibles a la irradiación. James Ewing, en su momento, opinó, en forma errónea, que el tumor era de origen endotelial. Aunque aún existe controversia en cuanto al origen de este tumor, el consenso actual es que este tumor se origina de los progenitores de la cresta neural, formando parte del espectro de los tumores neuroectodérmicos primitivos (PNET) que pueden ocurrir en tejidos blandos y en el sistema nervioso central. Hoy en día se reconoce que el SE forma parte de un espectro de neoplasias conocidos como familia de tumores de SE (FSE), el cual incluye tumores de células pequeñas de la pared costal (tumor de Askin) y el SE atípico (2,3,4).
En la mayoría de los SE han descrito una translocación cromosómica característica 11:22 al igual que en los otros PNET. Aproximadamente 5 % al 10 % de los casos tienen translocaciones alternas entre los cromosomas 21 y 22. La translocación 11:22 yuxtapone al gen EWS con el gen FLI, el cual es un miembro de la familia de los factores de transcripción ETS. La translocación 21:22 produce una proteína de fusión similar que combina el gen EWS con el factor de transcripción ERG, el cual es también un miembro de la familia ETS. El oncogén producto de la yuxtaposición EWS/FLI actúa como un factor de transcripción que puede producir transformación maligna en algunas líneas celulares. Es necesario conocer con mayor precisión los mecanismos de regulación de las diferentes vías de transmisión de la señal en los SE, un posible blanco terapéutico del oncogén EWS/FLI es el factor del crecimiento PDGF-C (5,6).
Los factores pronósticos del SE incluyen: sitio del tumor primario, volúmen de la enfermedad, extensión a tejidos blandos, y lo más importante la presencia de metástasis (7). La clasificación por estadios ha sido rudimentaria, con división en dos grupos: enfermedad localizada y metastásica. Dentro del grupo de pacientes con enfermedad localizada, los primarios localizados en las porciones distales de las extremidades, tienden a evolucionar más satisfactoriamente que los tumores centrales. Asimismo, el volúmen de la enfermedad y la extensión extra ósea se asocian con un pronóstico más desfavorable.
El tratamiento locorregional con cirugía o radioterapia sólo rara vez es curativo. Históricamente la sobrevida a los 5 años estaba en el orden del 10 % antes de la introducción del tratamiento sistémico efectivo. El desarrollo de la quimioterapia múltiple en conjunto con el tratamiento local para el tumor primario ha sido una historia exitosa en oncología, y ha permitido mejorar de manera significativa las cifras de sobrevida tanto en enfermedad localizada como en casos con metástasis a distancia.
El objetivo del presente trabajo es presentar nuestra experiencia en el manejo multidisciplinario del SE en pacientes menores de 18 años durante un período de quince años.
MÉTODO
Se realizó una revisión retrospectiva de las historias clínicas de los pacientes menores de 18 años con SE tratados en U.R.O.G y el S.R.L.T durante los años 2000 hasta el 2015. Los datos obtenidos de las historias clínicas fueron registrados en una hoja de cálculo en Microsoft Office Excel®. Para el seguimiento en casos necesarios se obtuvo la información contactando los pacientes y/o al médico referente por vía telefónica. Se obtuvo una estadística descriptiva de la población en cuanto a datos demográficos, presentación clínica, estudios diagnósticos, tratamiento, complicaciones, cifras de sobrevida y control local por el método actuarial Kaplan Meier (8).
Se incluyeron un total de 25 pacientes, la edad promedio fue de 10,8 años con un rango de edades de 1 a 18 años. De un total de 25 pacientes; 15 (68,1 %) pacientes fueron del sexo femenino y 10 (45,4 %) pacientes del sexo masculino.
Los SE se clasifican en enfermedad localizada o enfermedad metastásica asimismo, pueden presentarse a nivel óseo y extraóseo (1). De los 25 pacientes incluidos en el estudio 3 (12 %) corresponden a tumor de Askin todos presentaban enfermedad localizada. De los 22 pacientes restantes 15 (68,1 %) pacientes presentaban enfermedad localizada y 7 (31,8 %) pacientes enfermedad metastásica (Cuadro 1).

Cuadro 1
Distribución de acuerdo a localización y extensión
En cuanto, a la localización anatómica se pudo observar que 8 (32 %) de los pacientes el sitio primario fue a nivel de extremidades inferiores, 5 (20 %) pacientes de localización en pelvis y otros en columna, cabeza y cuello, tronco y extremidades superiores (Cuadro 2).

Cuadro 2
Distribución de acuerdo a la localización anatómica
La quimioterapia se administró en 25 pacientes con SE, el esquema de quimioterapia más frecuente fue vincristina, adriamicina, ciclofosfamida (VAC) alterno con ifosfamida, etopósido en 22 pacientes con enfermedad localizada y metastásica, un paciente con enfermedad ósea metastásica recibió como segunda línea de tratamiento temozolamida e irinotecan.
La radioterapia fue administrada en 24 pacientes, la paciente restante presentó un tumor a nivel del peroné enfermedad localizada, fue tratada con quimioterapia y resección completa de la lesión. El tratamiento radiante fue administrado mediante diferentes técnicas, con un acelerador lineal (AL) de energía dual empleando fotones de 6 y 18 MV siendo la más frecuente radioterapia conformada 3D en 14 pacientes (Figura 1 y 2), en 4 al principio del estudio se utilizaron técnicas convencionales 2D mediante un AL de 4 MV, en los últimos años del estudio 6 pacientes fueron tratados con técnicas de intensidad modulada y terapia modulada con arcos volumétricos.
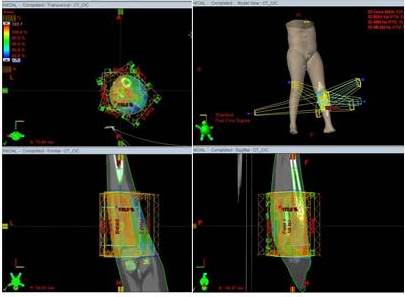
Figura 1
Paciente masculino de 11 años con SE extraóseo en región tibial izquierda enfermedad localizada. Se realizó biopsia incisional. QT: vincristina, doxorrubicina y ciclofosfamida con ifosfamida, etopósido. Luego RTIM a la semana 12, dosis 5 580 cGy, paciente actualmente VSE a los 52 meses. Plan de tratamiento en proyecciones axial, coronal y sagital, donde se aprecia la distribución porcentual de la dosis representada por diversas tonalidades de colores, cuyos valores pueden observarse en la columna de la izquierda. Posterior continuó con QT hasta cumplir 48 semanas.
La dosis de RT utilizada, por lo general, se basó en las recomendaciones del intergrupo de estudio del SE (IESSW). En las lesiones de huesos largos se irradia la lesión con un margen inicial de 5 cm hasta una dosis de 3 960 a 4 500 cGy, y luego reducción del campo al tumor con un margen de 2 cm hasta una dosis total de 5 580 cGy para enfermedad macroscópica, y 5 040 cGy para enfermedad microscópica. En las lesiones vertebrales no se debe exceder a 4 500 cGy debido a la tolerancia de la médula espinal. En los pacientes con enfermedad localizada las dosis oscilaron de 4 500 cGy a 5 680 cGy con fracciones de 180 cGy diarios. En los tres pacientes con tumor de Askin la dosis osciló de 4 600 cGy a 5 400 cGy con fracciones de 180- 200 cGy. En los pacientes con enfermedad metastásica las dosis variaron de 2 280 cGy a 6 140 cGy con fraccionamiento diario de 180 cGy, el paciente que recibió la dosis más alta presentaba un tumor gigante del húmero, por lo cual se administró una dosis mayor de forma concurrente con quimioterapia.
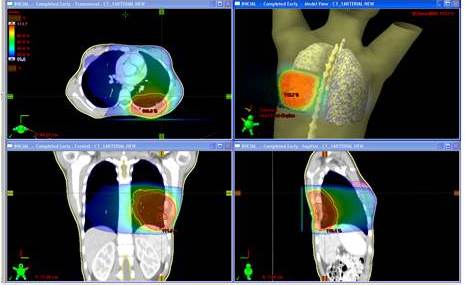
Figura 2
Paciente femenina de 11 años de edad con tumor de Askin en 8o arco costal izquierdo. Se realizó resección de 8o arco costal y resección de lóbulo inferior de pulmón izquierdo. Recibió quimioterapia con vincristina, doxorrubicina, ciclofosfamida alterno con ifosfamida, etopósido, luego en la semana 12 recibió RTC3D dosis 5 040 cGy y posteriormente QT hasta cumplir 48 semanas de tratamiento. Paciente actualmente VSE a los 38 meses. Plan de tratamiento en proyecciones axial, coronal y sagital, donde se aprecia la distribución porcentual de la dosis representada por diversas tonalidades de colores, cuyos valores pueden observarse en la columna de la izquierda.
El tiempo de seguimiento promedio fue de 42,5 meses con una mediana de 24,3 meses y un rango que osciló entre 1,1 a 180,9 meses; del total de 24 pacientes solo 3 se perdieron del control en diferentes períodos de tiempo.
RESULTADOS
Al analizar el total de los 25 pacientes con SE, los mismos se clasificaron en dos grupos SE localizado y SE metastásico, se encontró que 15 (61,5 %) presentaban enfermedad localizada de los cuales 7 fueron óseos y 8 extraóseos, existió una predominancia en las extremidades inferiores con 7 pacientes, paravertebrales y pelvis 3 pacientes cada uno, 2 a nivel de cabeza y cuello y 1 a nivel de tronco. Los 15 pacientes recibieron quimioterapia, siendo el esquema más utilizado en 10 pacientes el vincristina, doxorrubicina y ciclofosfamida (VAC) alterno con ifosfamida, etopósido. La RT fue administrada en 14 de los 15 pacientes, el paciente que no recibió RT presentaba un SE a nivel de peroné se administró quimioterapia y posterior resección completa de la lesión.
Al momento del estudio de los 15 pacientes SE localizado 8 (53,3 %) se encuentran vivos sin evidencia de enfermedad (VSE), y 5 (33,3 %) fallecieron a consecuencia de la enfermedad (MCE), 2 con recurrencias locorregional y MT a distancia y los 3 restantes no se pudo obtener información sobre el sitio de la recaída. Un paciente con tumor localizado en peroné tratado con quimioterapia y cirugía falleció 21 meses después del inicio del tratamiento a causa de una leucemia mieloide aguda sin evidencia de enfermedad de SE, y un paciente con SE paravertebral localizado se encuentra vivo con enfermedad, presentó recaída locorregional a los 14 meses de culminar la radioterapia.
Se registraron 3 pacientes con tumor de Askin, 2 de los pacientes se encuentran VSE a los 34 y 180 meses del tratamiento, y 1 paciente falleció a los 9 meses de finalizar tratamiento por desarrollar MT a nivel ósea.
En cuanto a los SE metastásicos se registraron 7 (26,9 %) pacientes todos de origen óseo, la localización anatómica predominante en estos pacientes fue en pelvis con 4 casos, 2 en extremidades inferiores y 1 en extremidad superior. Solo 1 de los 7 pacientes se encuentra vivo con enfermedad (VCE) con un seguimiento de 27 meses, los 6 pacientes restantes fallecieron a causa de la enfermedad en un período de tiempo de 9 a 35 meses.
Las cifras de sobrevida global (SG) actuariales de los 25 pacientes a los 5 años fue de 36,8 % con una desviación estándar (DE) 9,5 %; por otra parte, las cifras de sobrevida libre de enfermedad (SLE) a los 5 años fue de 34,4 % con una DE: 9,5 % (Figura 3).
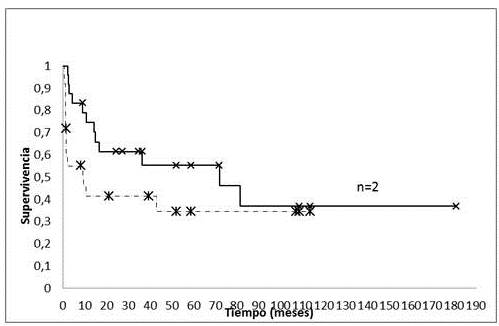
Figura 3
Sobrevida global y libre de enfermedad pacientes pediátricos con SE
La SG en los 18 pacientes con SE localizados fue de 75,6 % y 50,4 % a los 5 y 10 años respectivamente, con una media de tiempo de supervivencia de (112,3 ± 21,2) meses IC:95 % (70,8:153,8). De los 7 pacientes con SE metastásicos la SG a los 2 años fue de 28,6 %, con una media de tiempo de supervivencia de (17,4 ± 5,3) meses IC:9 5% (7,0 : 28,0) (Figura 4).
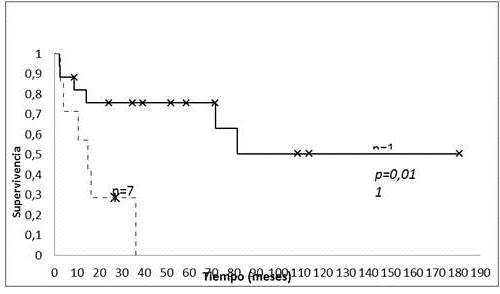
Figura 4.
Sobrevida global pacientes pediátricos con SE localizados vs., metastásicos.
La SLE a los 5 años para los pacientes con SE localizados fue de 48,7 %, con una media de tiempo libre de enfermedad de (61,8 ± 13,5) meses IC:95 % (34,8 : 87,6). Para los SE metastásica la SLE a los 2 meses fue de 14,3 %, con una media de tiempo libre de enfermedad de (2,5 ± 1,4) meses IC: 95% (0 : 5,1) (Figura 5).
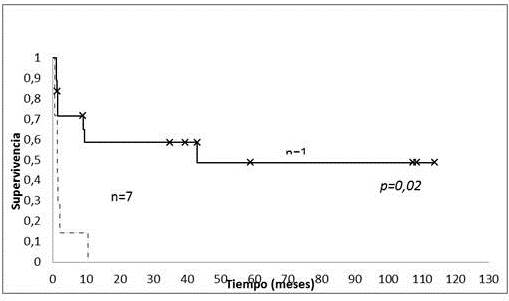
Figura 5
Sobrevida libre de enfermedad pacientes pediátricos con SE localizados vs., metastásicos
Las complicaciones observadas se presentan en el Cuadro 3. La toxicidad hematológica aguda, incluyó leucopenia en 9 pacientes solo uno de ellos grado IV, anemia en seis pacientes y trombocitopenia grado II en 1 paciente. La dermatitis fue frecuente en 15 pacientes, pero solo tres de ellos fue grado III. Las complicaciones gastrointestinales, usualmente náuseas y vómitos fueron moderadas, solo en cuatro pacientes, y en dos de ellos fue grado II. La toxicidad a nivel de mucosas se registró en solo un paciente y fue grado I, en ésta serie solo dos pacientes presentaron tumores en cabeza y cuello. Las complicaciones crónicas fueron poco frecuentes y se registraron en dos pacientes que presentaron fibrosis moderada de piel y tejido subcutáneo. 1 paciente con SE paravertebral localizado, presentó escoliosis severa que ha ameritado sucesivas cirugías ortopédicas. Otras complicaciones incluyeron azoospermia en un paciente secundario a quimioterapia. Dos pacientes desarrollaron neoplasias secundarias, un paciente con tumor de Askin desarrolló un condrosarcoma en la pared costal del campo de RT 15 años después del tratamiento con quimioterapia y RT, el cual fue tratado con cirugía y RT estando en la actualidad vivo sin enfermedad; el otro paciente presentó un tumor en peroné tratada con quimioterapia y cirugía desarrolló leucemia mieloide aguda muriendo por esta causa sin evidencia de recidiva del SE.
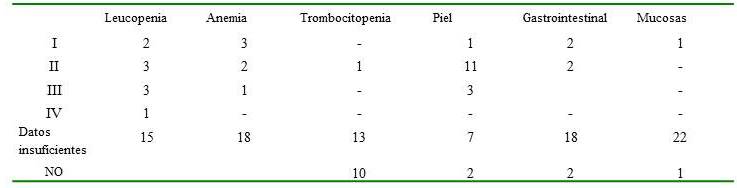
Complicaciones agudas durante el tratamiento
DISCUSIÓN
Los progresos obtenidos en el tratamiento del SE, se deben fundamentalmente, a los trabajos cooperativos de grupos interdisciplinarios a nivel internacional en los EE.UU y Europa. En la actualidad las cifras de sobrevida global a los 5 años en enfermedad localizada oscilan de 65 % a 75 % y en pacientes con metástasis menos del 30 %, excepto en pacientes con metástasis pulmonares aisladas en los cuales es alrededor de 50 % (9).
Los factores pronósticos del SE incluyen: sitio del tumor primario, volúmen de la enfermedad, extensión a tejidos blandos y la presencia de metástasis (7). Dentro del grupo de pacientes con enfermedad localizada, los tumores de las porciones distales de las extremidades, tienen una evaluación más satisfactoria que los tumores centrales. Asimismo, el volumen de la enfermedad y la afectación extraósea se asocian a un pronóstico más desfavorable (7).
Los tumores de la FSE deben ser considerados como una enfermedad sistémica desde el comienzo, aunque menos del 25 % de los pacientes tienen metástasis aparentes en el momento del diagnóstico inicial, el 80 % a 90 % de los casos van a desarrollar metástasis después de tratamiento local solamente, lo que hace pensar que la gran mayoría presentan enfermedad subclínica metastásica no aparente. Esto está en correlación con el hecho de que la reacción en cadena de la polimerasa de la transcriptasa reversa (RT-PCR) puede detectar enfermedad metastásica en casos de SE con enfermedad aparentemente localizada clínicamente (1).
El desarrollo de la quimioterapia (QT) en el SE ha sido continuo. Inicialmente se emplearon regímenes de 3 o 4 drogas vincristina, actinomicina D y ciclofosfamida (VAC) y vincristina, doxorrubicina, ciclofosfamida y actinomicina D (VDCA o VACA) observándose mejores resultados con el último esquema señalado (10). Luego se demostró que la actinomicina D podía ser omitida sin comprometer los resultados a largo plazo (11,12). Más recientemente se incluyó en el régimen de vincristina, doxorubicina, ciclofosfamida (VDC) ciclos alternos de ifosfamida, etopósido (IE) observándose mejoría en la sobrevida libre de recaídas de 5 años en pacientes con enfermedad localizada, al incluir estas dos drogas (13). Por tanto, en la actualidad el régimen estándar de quimioterapia incluye estos 5 agentes: vincristina, doxorrubicina, ciclofosfamida con ifosfamida, etopósido (VDC/IE) (13) .Por lo general, se administran en enfermedad localizada 4-6 ciclos de este esquema de QT antes del tratamiento local, y si hay respuesta adecuada se continúa con QT adicional después de la cirugía y o la RT. La respuesta inicial al tratamiento sistémico tiene importancia en el pronóstico. Se han empleado igualmente regímenes de QT más intensivos especialmente en pacientes con enfermedad avanzada o recaídas, los resultados de estos estudios son controversiales y el papel de estos esquemas intensivos aún no está establecido(9, 14).
El tratamiento local del SE, se puede realizar con cirugía, RT o ambas modalidades. Por lo general, se prefiere el tratamiento quirúrgico para evitar las secuelas tardías de la RT, siempre que éste no implique un compromiso funcional significativo. En huesos que pueden ser sacrificados, como el peroné, costillas y lesiones pequeñas de las manos y pies, es preferible la cirugía. Se ha empleado el papel de la resección quirúrgica con la utilización de técnicas que emplean injertos alogénicos de hueso o la colocación de prótesis articulares, las cuales pueden producir resultados funcionales favorables y facilitar la resección completa. En otras localizaciones donde no es posible la resección quirúrgica, o en lesiones extensas, la RT es el método de elección (1).
El SE, es un tumor radiosensible que puede ser controlado con dosis moderadas de RT. Luego de la QT inicial, en la semana 12 usualmente se administra el tratamiento local. La RT en enfermedad localizada puede ser utilizada como tratamiento local único, o en combinación con la cirugía, como tratamiento preoperatorio o posoperatorio, en tumores voluminosos en ciertas localizaciones como la pelvis, donde el tratamiento combinado puede permitir la realización de un procedimiento quirúrgico más limitado, mejorar el control local y obtener mejores resultados funcionales. Asimismo, la RT se emplea en pacientes con metástasis, en conjunto con la QT, lográndose una mejoría en la sobrevida libre de progresión y alivio de síntomas como el dolor (15). Los campos de tratamiento deben incluir el volúmen original del tumor con margen de 1,5 cm a 2 cm. La resonancia magnética es el método preferido para delinear la extensión del tumor antes de quimioterapia. En algunos casos puede ser preferible utilizar el volumen pos-quimioterapia para la planificación del tratamiento radiante.
La dosis prescrita típicamente es 5 580 cGy en 31 fracciones. Las lesiones localizadas en las vértebras deben tratarse con dosis de 4 500 cGy para respetar la tolerancia de la médula espinal. En caso de radioterapia posoperatoria después de la resección quirúrgica, las dosis oscilan entre 4 500 a 5 040 cGy, con campos que abarquen el lecho del tumor primario. La quimioterapia con ifosfamida y etopósido debe continuarse durante la radioterapia, evitando la utilización concurrente de doxorrubicina. En pacientes con enfermedad metastásica, es razonable considerar tratamiento radiante para las lesiones secundarias cuando sea posible. Convencionalmente, la radioterapia en estos casos debe utilizarse tardíamente en el transcurso del tratamiento sistémico de acuerdo a la respuesta. En caso de metástasis pulmonares la RT es de utilidad y mejora la sobrevida libre de recaídas (16,17,18,19,20,21,22). Las metástasis pulmonares a menudo se tratan con campos que abarquen ambos pulmones con dosis de 1 200 a 1 400 cGy, aunque existen ciertas controversias en este sentido. En presencia de enfermedad macroscópica pulmonar, se debe administrar una dosis adicional con campos reducidos hasta 4 500 cGy. En el caso de tumores de Askin, en los cuales se puede producir invasión de la pleura parietal y derrame pleural maligno, el empleo de radioterapia a dosis baja a todo el hemitórax, 1 200 a 1 400 cGy, produce una mejoría en la sobrevida libre de enfermedad reduciendo el riesgo de metástasis pulmonares subsecuentes (23). En la actualidad, se recomienda el empleo de radioterapia conformada con planificación 3D (RTC3D) y radioterapia con intensidad modulada (RTIM) para disminuir la toxicidad de los tejidos normales (24,25,26) En la presente serie se utilizó RTC3D en 13 (52 %) pacientes, y la RTIM en 6 (24 %) individuos
Tres casos clínicos han sido reportados en la literatura venezolana, un SE costal (27), un SE extraóseo de localización renal (28) y un SE paravertebral dorsal alto con cuadro clínico de compresión medular (29).
Los resultados obtenidos en la presente serie de 25 pacientes, en cuanto a sobrevida son inferiores a los reportados en los estudios de los grupos cooperativos antes mencionados (1). Debemos señalar sin embargo, que este es un análisis retrospectivo, donde los pacientes recibieron tratamiento sistémico en varias instituciones, y en muchos pacientes no se conocen los detalles de la quimioterapia (QT).
La terapéutica moderna, del SE, con fines curativos, emplea regímenes agresivos asociados con una toxicidad significativa, en ésta población de pacientes en edad pediátrica. Las complicaciones agudas usuales de la RT a nivel de piel y mucosas, tracto gastrointestinal, médula ósea se pueden exacerbar con el uso concomitante de QT. Algunos agentes como la doxorrubicina y la actinomicina D, no deben usarse concomitantemente con la RT. Las secuelas tardías del tratamiento con RT incluyen trastornos del crecimiento (30) efecto que podría reducirse al proteger las placas epifisarias en huesos largos. Asimismo, se debe siempre de tratar de proteger una franja de tejido sano en el tratamiento de tumores de las extremidades para reducir la fibrosis y el linfedema (31,32).
El empleo de técnicas modernas de RT, como la radioterapia conformada con planificación 3D y la radioterapia de intensidad modulada, pueden mejorar los resultados funcionales en tumores de las extremidades (32,33,34,35). Por otra parte, la aparición de neoplasias secundarias, representa otro problema a largo plazo, especialmente osteosarcoma inducidos por RT, y leucemias asociadas con los agentes alquilantes y las epidofilotoxinas (30,31,32,33,34,35), el riesgo de inducción de tumores secundarios depende de la dosis de radiación, y se pueden minimizar manteniendo dosis por debajo de los 6 000 cGy, en las series modernas la incidencia de sarcomas secundarios con megavoltaje está en el orden de 6 % a 7 % (35). En la presente serie, se encontró un caso de condrosarcoma del área irradiada, en un paciente con tumor de Askin a los 15 años después de tratamiento con QT y RT, y otra paciente con tumor en peroné tratada con QT y cirugía desarrolló leucemia mieloide aguda muriendo por esta causa sin evidencia de recidiva del SE.
REFERENCIAS
1. Roberts, Ruíz F, Ruan L. Tumores pediátricos en: Urdaneta N, Vera A, Peschel R, Wilson L. editores. Radioterapia Oncológica Enfoque Multidisciplinario. 2a edición. Venezuela: Disinlimed; 2009.p.1267.
2. Askin FB, Rosai J, Sibley RK, Dehner LP, McAlister WH. Malignant small cell tumor of the thoracic-pulmonary region in childhood: A distinctive clinic pathologic entity of uncertain histogenesis. Cancer. 1979;43:2438-2451.
3. Llombart-Bosch A, Lacombe MJ, Contesso G, Peydro-Olaya A. Small round blue cell sarcoma of bone mimicking atypical Ewing's sarcoma with neuroectodermal features. An analysis of five cases with immunohistochemically and electron microscopic support. Cancer. 1987;60:1570-1582.
4. Grier HE. The Ewing family of tumors. Ewing's sarcoma and primitive neuroectodermal tumors. Pediatr Clin North Am. 1997;44:991-1004.
5. May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, et al. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol. 1993;13:7393-7398.
6. Prieur A, Tirode F, Cohen P, Delattre O. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol Cell Biol. 2004;24:7275-7283.
7. Cotterill SJ, Ahrens S, Paulussen M, Jürgens HF, Voûte PA, Gadner H, et al. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol. 2000;18:3108
8. Matthews D, Farewell V. Using and understanding medical statistics. 4a edición. Nueva York: Karger; 2007.p.54.
9. Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, et al. Ewing Sarcoma: Current management and future approaches through collaboration. J Clin Oncol. 2015;33(27):3036-3046.
10. Nesbit ME Jr, Gehan EA, Burgert EO Jr, Vietti TJ, Cangir A, Tefft M, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing's sarcoma of bone: A long-term follow-up of the First Intergroup study. J Clin Oncol. 1990;8:1664-1674.
11. Burgert EO Jr, Nesbit ME, Garnsey LA, Gehan EA, Herrmann J, Vietti TJ, et al. Multimodal therapy for the management of no pelvic, localized Ewing's sarcoma of bone: Intergroup study IESS-II. J Clin Oncol. 1990;8:1514-1524.
12. Smith MA, Ungerleider RS, Horowitz ME, Simon R. Influence of doxorubicin dose intensity on response and outcome for patients with osteogenic sarcoma and Ewing's sarcoma. J Natl Cancer Inst. 1991;83:1460-1470.
13. Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003; 348:694-701.
14. Granowetter L, Womer R, Devidas M, Krailo M, Wang C, Bernstein M, et al. Dose-intensified compared with standard chemotherapy for non-metastatic Ewing sarcoma family of tumors: A Children's Oncology Group Study. J Clin Oncol. 2009;27:2536-2541.
15. Koontz BF, Clough RW, Halperin EC. Palliative radiation therapy for metastatic Ewing sarcoma. Cancer. 2006;106:1790-1793.
16. Paulussen M, Ahrens S, Craft AW, Dunst J, Fröhlich B, Jabar S, et al. Ewing's tumors with primary lung metastases: Survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol. 1998;16:3044-3052.
17. Dunst J, Paulussen M, Jürgens H. Lung irradiation for Ewing's sarcoma with pulmonary metastases at diagnosis: Results of the CESS-studies. Strahlenther Onkol. 1993;169:621-623.
18. Paulussen M, Ahrens S, Burdach S, Craft A, Dockhorn-Dworniczak B, Dunst J, et al. Primary metastatic (stage IV) Ewing tumor: Survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol. 1998;9:275-281.
19. Pilepich MV, Vietti TJ, Nesbit ME, Tefft M, Kissane J, Burgert EO, et al. Radiotherapy and combination chemotherapy in advanced Ewing's Sarcoma-Intergroup study. Cancer. 1981; 47:1930-1936.
20. Spunt SL, McCarville MB, Kun LE, Poquette CA, Cain AM, Brandao L, et al. Selective use of whole-lung irradiation for patients with Ewing sarcoma family tumors and pulmonary metastases at the time of diagnosis. J Pediatr Hematol Oncol. 2001; 23:93-98.
21. Cangir A, Vietti TJ, Gehan EA, Burgert EO Jr, Thomas P, Tefft M, et al. Ewing's sarcoma metastatic at diagnosis. Results and comparisons of two intergroup Ewing's sarcoma studies. Cancer. 1990;66:887-893.
22. Casey DL, Alektiar KM, Gerber NK, Wolden SL. Whole lung irradiation for adults with pulmonary metastases from Ewing sarcoma. Int J Radiat Oncol Biol Phys. 2014;89:1069-1075.
23. Schuck A, Ahrens S, Konarzewska A, Paulussen M, Fröhlich B, Könemann S, et al. Hemi thorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys. 2002;54:830-838.
24. Indelicato DJ, Keole SR, Shahlaee AH, Shi W, Morris CG, Marcus RB Jr. Definitive radiotherapy for Ewing tumors of extremities and pelvis: Long-term disease control, limb function, and treatment toxicity. Int J Radiat Oncol Biol Phys. 2008;72:871-877.
25. Kinsella TJ, Loeffler JS, Fraass BA, Tepper J. Extremity preservation by combined modality therapy in sarcomas of the hand and foot: An analysis of local control, disease free survival and functional result. Int J Radiat Oncol Biol Phys. 1983;9:1115-1119.
26. Jentzsch K, Binder H, Cramer H, Glaubiger DL, Kessler RM, Bull C, et al. Leg function after radiotherapy for Ewing's sarcoma. Cancer. 1981;47:1267-1278.
27. Tovar I, Parra M, Villalón M, Cermeño T, Valle M, Garrido L. Sarcoma de Ewing costal. Neumol Pediatr. 2013;8 (1):39-43.
28. Godoy A, Liuzzi J, Parada D, Herrera J, Machado T, Velásquez Y. Tumor neuroectodérmico primitivo renal. Rev Venez Oncol. 2009;21(4):240-243.
29. Ferre Y, Suárez O, Muñoz D, Hernández PA, Olmedo FO, Noguera HO, et al. Sarcoma de Ewing: Variedad neuroepitelioma periférico con curso clínico de síndrome de compresión medular. Rev Mex Neuroci. 2010;11(5):368-372.
30. Thomas PR, Perez CA, Neff JR, Nesbit ME, Evans RG. The management of Ewing's sarcoma: Role of radiotherapy in local tumor control. Cancer Treat Rep. 1984;68:703-710.
31. Fuchs B, Valenzuela RG, Inwards C, Sim FH, Rock MG. Complications in long-term survivors of Ewing sarcoma. Cancer. 2003;98:2687-2692.
32. Tefft M, Lattin PB, Jereb B, Cham W, Ghavimi G, Rosen G, et al. Acute and late effects on normal tissues following combined chemo- and radiotherapy for childhood rhabdomyosarcoma and Ewing's sarcoma. Cancer. 1976;37:1201-1217.
33. Kuttesch JF Jr, Wexler LH, Marcus RB, Fairclough D, Weaver-McClure L, White M, et al. Second malignancies after Ewing's sarcoma: Radiation dose-dependency of secondary sarcomas. J Clin Oncol 1996; 14:2818-2825.
34. McLean TW, Hertel C, Young ML, Marcus K, Schizer MA, Gebhardt M, et al. Late events in pediatric patients with Ewing sarcoma/primitive neuroectodermal tumor of bone: The Dana-Farber Cancer Institute/Children's Hospital experience. J Pediatr Hematol Oncol. 1999;21:486-493.
35. Smith LM, Donaldson SS. Incidence and management of secondary malignancies in patients with retinoblastoma and Ewing's sarcoma. Oncology (Williston Park). 1991;5:135-141.
Notas de autor
fruiz2602@gmail.com.