Rabdomiosarcoma tratamiento multidisciplinario análisis de 27 pacientes
Rabdomiosarcoma tratamiento multidisciplinario análisis de 27 pacientes
Revista Venezolana de Oncología, vol. 30, núm. 3, pp. 175-186, 2018
Sociedad Venezolana de Oncología

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 4.0 Internacional.
Recepción: 23 Noviembre 2017
Revisado: 04 Marzo 2018
Aprobación: 12 Mayo 2018
Resumen: OBJETIVO: Presentar la experiencia en 27 pacientes menores de 18 años con diagnóstico de rabdomiosarcoma quienes recibieron tratamiento multidisciplinario. MÉTODO: Revisión retrospectiva de 27 pacientes tratados con cirugía, quimioterapia y radioterapia en el período 2000-2015. 22 presentaban diagnóstico inicial de rabdomiosarcoma y 5 recidivas. 14 (63,6 %) sitios desfavorables, Grupo de riesgo 10 (45,4 %) IIIA, 5 (22,7 %) IV. Estadio II 6 (27,2 %), estadio III- IV 5 (22,7 %). Riesgo intermedio 9 (40,9 %), riesgo bajo 8 (36,3 %). Todos recibieron quimioterapia, a 2 se les realizó resección completa. 20 recibieron radioterapia, RTC3D en 9, VMAT-RTIM en 7 y RT2D en 3. La dosis varió de 3 600-5 400 cGy. RESULTADOS: 22 con diagnóstico inicial rabdomiosarcoma: 12 MCS, 1 vivo con enfermedad, 9 vivos sin enfermedad, 1 paciente perdido. La SG a los 5 años 42,1 % y la SLE a los 5 años 36,4 %. En los grupos de riesgo I-II la SG a 5 años 50 % y grupo III-IV 36,8 %. De los 5 pacientes con recidivas, 3 vivos sin enfermedad, 1 perdido, 2 muertos con enfermedad. Las complicaciones agudas más importantes fueron toxicidad hematológica, dermatitis, mucositis y fibrosis crónica grado 2. CONCLUSIONES: Resultados obtenidos no son comparables con otras series. La mayoría de los pacientes fueron sitios desfavorables, enfermedad voluminosa y grupo III-IV. Problemas socio económico del país, cumplimiento inadecuado del tratamiento en el tiempo, explican en gran parte resultados desfavorables en esta serie.
Palabras clave: Cáncer, rabdomiosarcoma, tratamiento, radioterapia, quimioterapia, cirugía, sobrevida.
Abstract: OBJECTIVE: To report the experience in 27 patients under 18 years old with RMS who received multidisciplinary treatment. METHOD: Retrospective review of 27 patients with RMS treated with surgery, chemotherapy and radiotherapy in the period 2000-2015. 22 with initial diagnosis of rabdomiosarcoma and 5 has recurrent diseases. 14(63.6 %) has unfavorable sites. Risk groups: IIIA: 10(45.4 %) IV 5 (22.7 %). Stage II 6 (27.2), stage III- IV 5 (22.7) and 9(40.9 %) in intermediate risk, 8 patients (36.3 %) in low risk. All patients received chemotherapy, 2 had wide local resection. 20 received radiotherapy, 3DCRT in 9, VMAT-IMRT in 7 and 2DRT in 3, dose varied from 3 600-5 400 cGy. RESULTS: Of 22 patients with primary rabdomiosarcoma: 12 were dead with disease, 1 alive with disease, 9 alive without of disease and 1 lost of follow up. The 5 years OS was 42.1 % and the 5 years EFS 36.4 %. In risk group I-II 5 years OS was 50 % and in group III-IV 36.8 %. 5 patients with recurrent disease, 3 are alive without disease, 1 lost of follow up and 2 dead with disease. Complications included hematological toxicity, dermatitis, mucositis, and grade 2 with chronic fibrosis. CONCLUSIONS: These results are not comparable with others series. Most patients had tumors in unfavorable sites, bulky disease and group III. Socio economic problems in our country and inadequate compliance with treatment, largely explain unfavorable results in this series.
Keywords: Cancer, rhabdomyosarcoma, treatment, radiotherapy, chemotherapy, surgery, survival.
INTRODUCCIÓN
El rabdomiosarcoma (RMS) es un tumor agresivo originado de las células embrionarias del mesénquima con potencial de diferenciación a musculo estriado. Fue descrito inicialmente por Weber en 1854. Es una neoplasia rara de comportamiento clínico y biológico heterogéneo. Es el tumor de partes blandas más común en edad pediátrica, representando alrededor de 3 % a 4 % de todas las neoplasias en este grupo etario. Aproximadamente en EE.UU se diagnostican 350 casos nuevos por año siendo la incidencia anual de 4,3 casos por millón en menores de 20 años (1).
La mayoría de los casos de RMS son esporádicos, sin embargo, se ha encontrado asociación de la enfermedad con síndromes hereditarios como: la neurofibromatosis, síndromes de Li- Fraumeni, Beckwith- Wiedemann y Costello (2,3). Se han reportado cambios genéticos translocaciones que afectan el gen FKHR en el cromosoma 13, con fusión con el gen PAX7 y PAX3 en el RMS alveolar. En el RMS embrionario se ha encontrado pérdida de heterozigocidad en el 11p15.5 lo que sugiere la presencia de un gen supresor de tumor (4,5,6,7). En el futuro es probable que se desarrolle una clasificación molecular en base a la expresión de genes que pueda correlacionarse con la evolución clínica de la enfermedad y pueda ser de utilidad en el manejo terapéutico.
El RMS puede originarse en cualquier localización anatómica donde exista músculo esquelético, las áreas más frecuentes incluyen el sistema genitourinario y el área de cabeza y cuello. Entre los factores pronósticos además del tipo histológico y la extensión de la enfermedad, debe tomarse en cuenta la localización del tumor relacionado con la agresividad del mismo y la evolución clínica.
Dada la baja frecuencia de este tumor, se hizo necesaria la realización de estudios multiinstitucionales para desarrollar nuevos esquemas terapéuticos multidisciplinarios. Se han organizado grupos cooperativos en los (EE.UU) el de estudio del rabdomiosarcoma (IRSG) y en Europa la sociedad internacional de oncología pediátrica (SIOP), el trabajo multidisciplinario de estas y otras organizaciones, al integrar la cirugía, radioterapia y quimioterapia ha permitido mejorar las cifras de sobrevida y curación de 15 % a 25 % a principios del siglo XX y a más de del 70 % en la actualidad (4).
El objetivo del presente trabajo es reportar la experiencia de quince años en el manejo terapéutico del RMS, se hará énfasis en los aspectos relacionados con la radioterapia y quimioterapia.
MÉTODO
Se realizó una revisión retrospectiva de las historias clínicas de los pacientes menores de 18 años con RMS tratados en la unidad de radioterapia oncológica GURVE (U.R.O.G) y el servicio de radioterapia la Trinidad “Dr. Enrique M. Gutiérrez”(S.R.L.T) durante los años 2000 hasta el 2015. Se realizó una revisión de 35 historias clínicas, excluyendo siete pacientes que no recibieron quimioterapia (QT) ni radioterapia (RT) en nuestros servicios, quedando un total de 27 pacientes para el estudio; 23 individuos presentaban diagnóstico inicial de RMS de ellos uno fue excluido por solo recibir una dosis de 180 cGy no continuando la radioterapia quedando 22 pacientes, y los otros 5 fueron referidos para tratamiento por recaídas de la enfermedad habiendo sido tratados previamente en otros centros. Los datos obtenidos de las historias clínicas fueron registrados en una hoja de cálculo en Microsoft Office Excel®.
Para el seguimiento en casos necesarios se obtuvo la información contactando los pacientes y/o al médico referente por vía telefónica. Se obtuvo una estadística descriptiva de la población en cuanto a datos demográficos, presentación clínica, estudios diagnósticos, tratamiento, complicaciones, cifras de sobrevida y control local por el método actuarial Kaplan Meier (8).
En cuanto a la edad, el rango fue de 0,9-17 años con una edad promedio de 7,7 años. La distribución de acuerdo al género mostró un ligero predominio en el sexo masculino 15 pacientes (55,6 %) vs. 12 (44,4 %) pacientes del sexo femenino.
La clasificación histopatológica se presenta en el Cuadro 1. El RMS embrionario fue el grupo más frecuente en 78 % de los pacientes.

Cuadro 1
Distribución de los pacientes de acuerdo a la histopatología.
Los 22 pacientes referidos con diagnóstico inicial de RMS fueron clasificados de acuerdo a la localización anatómica en grupos de pronóstico favorable y desfavorable (Cuadro 2). Se observó un predominio de pacientes en las localizaciones anatómicas desfavorables 63,6 % de los casos.

Cuadro 2
Distribución de los pacientes de acuerdo a la localización anatómica
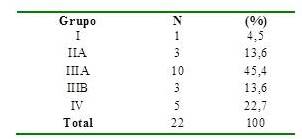
La clasificación por estadios de acuerdo a los grupos de riesgo del IRS, para los 22 pacientes con diagnóstico inicial de RMS se especifica en el Cuadro 3, siendo el grupo IIIA el de mayor número de casos para un total de 10 pacientes (45,4 %). Se analizan por separado los pacientes referidos por recaídas de la enfermedad, 5 individuos: a. 2 con tumores localizados inicialmente en cabeza y cuello grupo de riesgo inicial IIA y IIIA, y 1 con RMS de localización genitourinario grupo inicial IA, quienes presentaron recaídas
locorregionales, y b. 2 pacientes, uno con enfermedad en tórax grupo de riesgo inicial IA y el otro en extremidad inferior grupo de riesgo inicial IIIA respectivamente con metástasis a distancia.
Cuadro 3
Sistema de clasificación por grupos del IRS.
Asimismo, se clasificaron los pacientes de acuerdo al sistema TNM de la SIOP (9) el cual toma en cuenta el tamaño tumoral menor o mayor de 5 cm, la localización anatómica, afectación ganglionar y enfermedad a distancia, evidenciándose que de los 22 pacientes hubo predominio en los estadios II como se muestra en el Cuadro 4.

Cuadro 4
Distribución por estadios
De acuerdo a la clasificación por grupo de estratificación de riesgo 9 de los 22 pacientes con diagnóstico inicial de RMS 8 (36,3 %) fueron clasificados de bajo riesgo, 9 (40,9 %) riesgo intermedio y 5 con metástasis a distancia como alto riesgo.
La evaluación inicial de los pacientes al ingreso de los servicios de RT incluyó usualmente perfil de laboratorio, tomografía axial computarizada (TAC) con protocolo de RT para planificación del tratamiento radiante. En relación al tratamiento quirúrgico, en 14 pacientes solo se practicó biopsia de la lesión, en 1 paciente resección completa (lesión retroauricular), en 7 pacientes se realizó resección parcial de los cuales 3 tuvieron márgenes microscópicos positivos (R1) (lesiones en antebrazo, paratesticular, codo) y márgenes macroscópicos positivos (R2) en dos casos (lesiones en conducto auditivo y glúteo) y en 2 pacientes se desconoce el estado de los márgenes.
La RT fue utilizada en 20 de los 22 pacientes con diagnóstico inicial de RMS, se omitió la irradiación en un paciente con lesión en región retro auricular que fue resecada por completo y recibió luego QT, y en un paciente con RMS botrioide de la vagina tratada con QT y luego resección completa de la lesión con márgenes negativos. En los 5 pacientes con RMS con recaída se administró RT. En cuanto a la técnica de RT utilizada, los pacientes fueron tratados en un acelerador lineal (AL) de 4 MEV inicialmente y posteriormente en AL de energía dual empleando fotones de 6 y 18 MEV. Al principio se emplearon técnicas convencionales y planificación 2D en 3 (13,6 %) pacientes, posteriormente RT conformada con planificación 3D (RTC3D) y a partir del año 2004 esta fue la modalidad más empleada en 9 (40,9 %) pacientes (Figura 1), en un paciente se utilizaron técnicas 2D y 3D, y la RT de intensidad modulada con técnica de ventana deslizante (IMRT) y terapia modulada con arcos (VMAT-RAPIDARC) se empleó en 7 (31,8 %) pacientes.
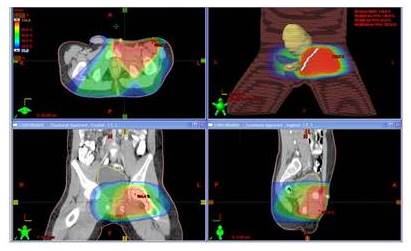
Figura 1
Paciente masculino de 12 años con RMS embrionario paratesticular izquierdo Grupo II, estadio IB. Se practica orquidectomía radical izquierda por vía inguinal, bordes de resección positivos. Recibe QT con VAC 3 ciclos, luego RTC3D a hemiescroto y región inguinal izquierdo, dosis 4 140 cGy, Plan de tratamiento en proyecciones axial, coronal y sagital, donde se aprecia la distribución porcentual de la dosis representada por diversas tonalidades de colores, cuyos valores pueden observarse en la columna de la izquierda. QT posterior hasta completar 37 semanas. Recidiva a nivel paraórtico a los 29 meses post tratamiento. Recibe QT con ifosfamida, carboplatino y etopósido sin respuesta adecuada. Se realiza resección con bordes positivos, se administra RT 4 500 cGy a región paraórtica, y posterior QT, paciente fallece por enfermedad retroperitoneal pero con control local a los 47 meses.
La dosis de RT utilizada, por lo general, se basó en las recomendaciones del IRSG, usualmente se emplearon fracciones diarias de 180 cGy cinco veces por semana hasta una dosis total de 3 600 a 4 100 cGy para enfermedad microscópica residual y de 4 500 cGy a 5 040 cGy para enfermedad macroscópica. La dosis administrada en el 90 % de los 20 pacientes con diagnóstico inicial de RMS estuvo dentro de estos rangos.
La QT fue administrada por nosotros en 12 pacientes, el resto recibieron el tratamiento sistémico en otros centros e instituciones públicas y privadas y fueron referidos a nuestro servicio para el tratamiento radiante. En 16 de 20 pacientes se utilizó el esquema actinomicina D, vincristrina, ciclofosfamida (VAC), tres pacientes recibieron etopósido y adriamicina, en ocho pacientes no fue posible encontrar datos sobre los agentes de quimioterapia utilizados.
El promedio de seguimiento en los 22 pacientes con diagnóstico inicial de RMS fue de 51,3 meses con una mediana de 32,8 meses, rango de 1,0 -178,3 meses.
RESULTADOS
Al analizar el estado de los pacientes en el último control se encontró que de los 22 pacientes con diagnóstico inicial de RMS, 12 pacientes fallecieron a consecuencia de la enfermedad (MCE), 1 está vivo con enfermedad (VCE) y 9 están vivos sin evidencia de enfermedad (VSE); se debe señalar que 1 de los 22 fue perdido de control después de finalizar RT. La sobrevida global (SG) a los 5 años de los 22 pacientes con diagnóstico inicial de RMS fue de 42,1 % a los, siendo la sobrevida libre de enfermedad (SLE) a los 5 años 36,4 % (Figura 2)
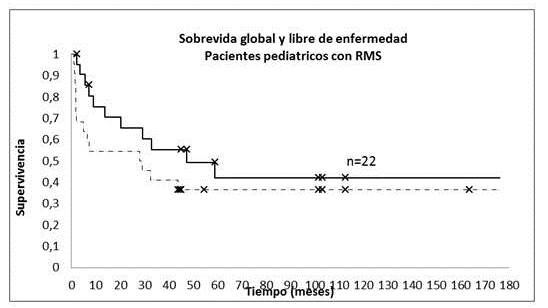
Figura 2
Al dividir los 22 pacientes por grupo de riesgo del IRS, se establecieron dos categorías: grupo de riesgo I-II vs. III-IV, calculándose las cifras de sobrevida, la SG a los 5 años fue de 50 %, con un error estándar en el primer caso elevado por el tamaño de la población, y para el grupo de riesgo III-IV la SG a los 5 años fue de 36,8 % (Figura 3).
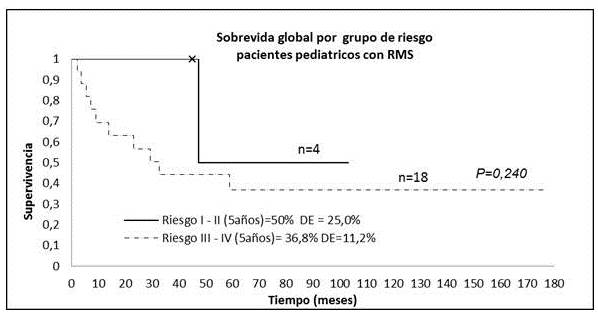
Figura 3.
En el grupo I-II solo 1 paciente con RMS paratesticular falleció por recaída a nivel de región paraórtica, los 3 restantes estan VSE. De los 13 pacientes en el grupo III , 6 estan MCE, y 6 pacientes se encuentran VSE, 1 paciente esta VCE. En cuanto a los 5 pacientes con RMS grupo IV todos estan MCE en un período de 2 a 29 meses después de finalizar el tratamientro.
En relación a los 20 pacientes con diagnóstico inicial de RMS tratados con RT, en 7 pacientes no se logró remisión de la enfermedad, se observaron recaídas locorregionales en 3 (15 %) pacientes, 4 pacientes presentaron metástasis (MT) a distancia en columna, retroperitoneo, sistema nervioso central (SNC) y pulmón.
En cuanto a los 5 pacientes referidos por recidivas de la enfermedad, 3 pacientes están VSE uno de ellos se perdió de control después de finalizar tratamiento habiendo logrado remisión de la enfermedad, y los otros dos con un tiempo de sobrevida de 22,5 meses y 178,3 meses. Dos pacientes fallecieron por causa de la enfermedad.
Las complicaciones observadas se presentan en el Cuadro 5, clasificadas por grado (10). La toxicidad hematológica aguda, incluyó leucopenia en 7 pacientes solo uno de ellos grado IV, anemia en cinco pacientes, y 3 pacientes con trombocitopenia, en uno de ellos grado III. La dermatitis fue frecuente en 16 pacientes, pero solo en 3 de ellos fue grado III. La toxicidad a nivel de mucosas se registró en 6 pacientes, en un 1 de ellos fue grado III. En cuanto a las complicaciones crónicas, solamente en un paciente se encontró fibrosis grado II, no se encontraron neoplasias secundarias.

Cuadro 5.
Complicaciones agudas
DISCUSIÓN
En la actualidad los fundamentos del tratamiento del RSM se basan en el concepto de terapia adaptada al grupo de riesgo, es fundamental la presencia de un equipo multidisciplinario en el manejo de estos pacientes, pues los resultados más favorables se han obtenido a través de la integración del tratamiento local cirugía y RT, con el tratamiento sistémico, pilar fundamental de la terapéutica del RMS. El tratamiento de esta afección ha evolucionado de manera significativa en los últimos años desde 1972 cuando se organiza el grupo de estudio del IRSG. Hasta el momento este grupo ha realizado cinco estudios cooperativos, los cuales han sido fundamentales para identificar los factores pronósticos, desarrollar esquemas terapéuticos adaptados al grupo de riesgo y mejorar los resultados de curación. Las cifras de sobrevida a los 5 años se han modificado significativamente de 55 % en el estudio IRSG-I a 75 % aproximadamente en los estudios IRSG- III y IRSG- IV (11,12). En la actualidad el IRSG se integró al comité de sarcomas de partes blandas del grupo oncológico infantil (COG), y se han realizado protocolos de investigación ARST 0331 para pacientes con RMS riesgo bajo, ARST0531 para riesgo intermedio y ARST0431 y ARST08P1 para pacientes de alto riesgo (13,14,15,16,17).
La cirugía constituye una modalidad importante del tratamiento de la enfermedad localizada, siempre que se pueda realizar una resección completa de la enfermedad con resultados cosméticos y funcionales aceptables, en caso de irresecabilidad, o en tumores a nivel de órbita, vagina, o tracto biliar, es preferible la realización de una biopsia seguida de QT neoadyuvante y posteriormente tratamiento local definitivo. Los tumores de cabeza y cuello rara vez son susceptibles a una resección completa. En las lesiones de las extremidades siempre que sean posibles se debe intentar la resección completa sino se compromete la función del miembro, igualmente se recomienda la realización de ganglio centinela (18,19). En los tumores genitourinarios, exceptuando los tumores paratesticulares, la cirugía inicial conlleva a una morbilidad significativa por lo cual se recomienda tratamiento de inducción con QT y de acuerdo a la respuesta tratamiento quirúrgico. Los tumores paratesticulares forman un grupo diferente y deben ser tratados con orquidectomía inguinal radical, y en los pacientes mayores de 10 años, en la actualidad, se recomienda la realización de una disección retroperitoneal con preservación de los plexos nerviosos, porque en este grupo de pacientes sin evidencia de alteraciones en los estudios de imágenes, existe un riesgo cerca del 50 % de metástasis ganglionares a este nivel, lo que hace necesario el tratamiento con QT más intensiva y RT (20,21). La cirugía también ha sido utilizada en el tratamiento de la enfermedad metastásica, y sus indicaciones deben ser individualizadas.
La RT constituye una modalidad terapéutica fundamental, en particular para lograr el control local de la enfermedad residual microscópica o macroscópica después de cirugía y QT, en la actualidad se recomienda su uso en todos los pacientes excepto aquellos RMS embrionarios grupo I. El RMS es uno de los sarcomas de partes blandas más sensibles a las radiaciones. Las áreas de enfermedad macroscópica en el RMS deben ser tratadas con dosis de 5 040 cGy y en enfermedad microscópica 4 140 cGy, utilizando fracciones diarias de 180 cGy.
La respuesta a la QT se ha utilizado como criterio para determinar si la dosis de RT puede reducirse en pacientes con buena respuesta al tratamiento sistémico (22), las pautas del IRSV permiten una reducción de la dosis a 3 600 cGy en pacientes con estadio I, con histología alveolar o indiferenciado y en otros pacientes con estadio II (enfermedad microscópica residual). En los pacientes del grupo 3 con primario de la órbita o del párpado una dosis de 4 500 cGy es efectiva y no excede la tolerancia de la retina. En el estudio IRS-V se ha recomendado, en pacientes seleccionados, el empleo de una nueva intervención quirúrgica, sin embargo, en el RMS grupo 3 la resección completa en la semana 12 no puede obviar el empleo del tratamiento radiante, pero se permite una reducción de la dosis a 3 600 cGy si en la intervención quirúrgica subsecuente el tumor pudo ser resecado con márgenes negativos y 4 140 cGy en casos con márgenes quirúrgicos positivos; en todos los otros casos la dosis estándar para el grupo 3 es de 5 040 cGy, en pacientes con enfermedad metastásica la RT puede ser utilizada para controlar el primario y las áreas de depósitos secundarios, la dosis recomendada es 5 040 cGy, excepto en la órbita 4 500 cGy.
El volúmen de tratamiento se ha ido reduciendo, a medida que se conoce con mayor precisión la extensión de la enfermedad por medio de los estudios de imágenes. El volúmen tumoral macroscópico se define en base a la extensión de la enfermedad prequimioterapia. En el pasado se empleó comúnmente un margen de 2 cm, las recomendaciones actuales del IRS señalan un margen de 1,5 cm. En el IRS-V se permite igualmente una reducción del volúmen de tratamiento a los 3 600 cGy para utilizar un margen de 5 mm alrededor del GTV si no existía diseminación linfática en el momento del diagnóstico inicial. En pacientes con ganglios positivos, la reducción del volúmen se debe realizar a los 4 140 cGy. En la actualidad, se recomienda el empleo de RT conformada con planificación 3D (RTC3D) y RT con intensidad modulada (RTIM) para disminuir la toxicidad de los tejidos normales (23). En la presente serie se utilizó RTC3D en 9 (40,9 %) pacientes, y la RTIM en 7 (31,8 %) individuos, la braquiterapia puede ser igualmente una opción adecuada en pacientes con tumores superficiales o en algunas localizaciones en la pelvis.
La QT constituye una modalidad fundamental, el tratamiento sistémico debe ser empleado en todos los pacientes. En la actualidad el esquema estándar en el RMS continua siendo vincristina, actinomicina D y ciclofosfamida (VAC) (24). Los pacientes con enfermedad favorable pueden ser tratados con dos drogas, eliminando la ciclofosfamida (24,25). Se han realizado estudios para evaluar la utilidad de otras drogas como la doxorrubicina, no observándose un beneficio en pacientes con riesgo intermedio. Por otra parte, en pacientes con riesgo intermedio se comparó el VAC con VAI y el VIE (I: Ifosfamida, E: etopósido), sin observarse un beneficio al comparar estos esquemas con el VAC estándar (26). En los pacientes con enfermedad avanzada se han utilizado otras drogas como DTIC, adriamicina, cisplatino y topotecan, en particular este último agente ha sido ensayado en el IRS-V en pacientes con riesgo intermedio (27). En la actualidad se están desarrollando régimen de QT intensiva con ifosfamida/etopósido y vincristina, doxorrubicina, ciclofosfamida e irinotecan en pacientes con alto riesgo (28).
La QT se continúa durante la RT, pero se omite la actinomicina D durante el tratamiento radiante para prevenir el aumento de las reacciones agudas observadas con la administración concurrente de esta droga. Actualmente la RT se introduce en la semana 12 del tratamiento, después de 4 ciclos de QT. En los pacientes con enfermedad parameníngea en los cueles exista extensión intracraneal evidente, tradicionalmente la RT se ha indicado al principio del tratamiento, sin embargo, en estudios recientes han señalado que la RT podría ser administrada con una secuencia similar a la antes mencionada. De acuerdo a las recomendaciones de los protocolos ARST0331 para pacientes de bajo riesgo se inicia la RT en la semana 13, en el ARST0531 para pacientes riesgo intermedio en la semana 4, y para los pacientes de alto riesgo con MT, la RT se permite en la semana 20-25, pero puede utilizarse en la semana 1-6 cuando exista infiltración paraespinal o intracraneal, y en la semana 47-52 para enfermedad metastásica extensa (28).
El estadio, histología y tratamiento de la enfermedad están relacionados al pronóstico. Para pacientes del grupo de bajo riesgo la SG a largo plazo es alrededor de 90 %, el grupo intermedio 70 % y pacientes de alto riesgo 20 %. El control local (CL) está más relacionado con el sitio de enfermedad primaria que con la histología en pacientes grupo 3 que se le administró RT (29,30,31). Los estudios del IRS reportan tasas de CL de 95 % en tumores de las extremidades y en cabeza y cuello en sitios no parameníngeos (32), por su parte, para aquellos de localización parameníngeas el control de la enfermedad es alrededor del 85 % y en los otros sitios es alrededor del 90 % (33,34,35). Es importante resaltar que el RMS alveolar aun cuando la enfermedad sea totalmente resecada, se requiere RT (35).
Donaldson y col., presentaron una comparación de los resultados obtenidos por el grupo europeo (SIOP) con el estudio tumores mesenquimales malignos (MMT 89) en el cual se trató de omitir la RT inicialmente y se prefirió el uso de la cirugía como tratamiento local, empleando la irradiación solo después de resección incompleta, afectación ganglionar o una respuesta desfavorable a la QT vs., el IRSG IV en el cual se utilizó RT en todos los pacientes con resección incompleta, histología alveolar y en todos los pacientes con enfermedad parameníngea, observándose una mejoría tanto en la SG como en la sobrevida libre de recaída (SLR) con el protocolo del IRSG IV, como se muestra en el Cuadro 6, este hecho resalta la importancia del uso de la RT como parte del tratamiento combinado en los pacientes con RMS (34) .
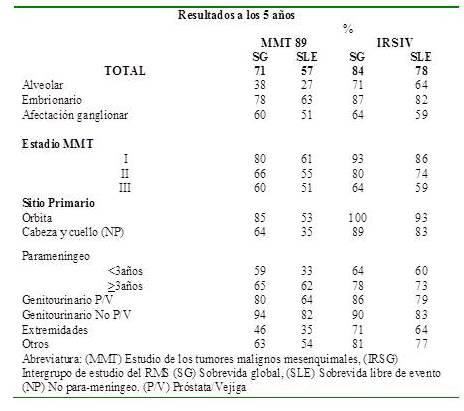
Cuadro 6
Los resultados en los 22 pacientes con diagnóstico inicial de RMS de esta serie indican que 54,5 % de ellos fallecieron a causa de la enfermedad, 1 paciente se encuentran vivo con enfermedad y 9 pacientes están vivos sin enfermedad. La sobrevida global (SG) a los 5 años de los 22 pacientes con diagnóstico inicial de RMS fue de 42,1 %, siendo la sobrevida libre de enfermedad (SLE) 36,4 %.
Es importante resaltar que el presente trabajo se hizo en base a un estudio retrospectivo, el cual tiene ciertas limitaciones. De los 22 pacientes con diagnóstico inicial de RMS 13 recibieron QT en diferentes centros, en muchos de ellos se desconoce los detalles en cuanto a la administración regular y adecuada del tratamiento sistémico, de igual manera, este grupo de pacientes no inició la RT según los estándares internacionales, hecho que tiene mayor relevancia dado a la situación de deterioro de la atención médica del país. Además muchos de los pacientes en estas series presentaban factores desfavorables: histología alveolar 13,7 %, localización anatómica desfavorable 63,4 %, enfermedad avanzada grupos III y IV 81,8 % de los casos, enfermedad voluminosa 45,4 %. Estos hechos explican en gran parte los resultados obtenidos en cuanto a sobrevida, que son inferiores a los obtenidos en los estudios cooperativos internacionales antes mencionados.
En una revisión retrospectiva de pacientes con RMS del año 2007 en el Hospital J.M de los Ríos, servicio de oncología 59 pacientes recibieron QT, 41 de manera regular y 18 pacientes de manera irregular. Solo 31 pacientes recibieron RT, 28 con fines curativos y 3 con fines paliativos. Estos autores reportan un total de 27 pacientes vivos (45,7 %), 24 sin enfermedad y 3 con enfermedad (35).
La terapéutica multidisciplinaria del RMS se asocia a secuelas significativas a largo plazo, muchas de ellas asociadas con el uso de la RT y/o QT, además de la mutilación quirúrgica en caso de que requiera cirugía radical. Entre los efectos secundarios tardíos incluyen: retardo de crecimiento, problemas dentales, alteraciones visuales (36,37,38,39). Igualmente se han reportado en tumores genitourinarios problemas de incontinencia urinaria, aumento de la frecuencia nocturna, cistitis hemorrágicas, así como problemas intestinales (36). En pacientes que reciben altas dosis de ciclofosfamida se han encontrado aumento de riesgo de daño cardíaco. Asimismo, existe un riesgo de endocrinopatías después de la irradiación de RMS en cabeza y cuello. Otros de los problemas preocupantes es el desarrollo de tumores secundarios, los cuales son más frecuentes en pacientes tratados con RT y QT, especialmente sarcomas óseos y leucemias mieloides agudas (LMA), 22 casos en 1 770 pacientes en los pacientes tratados en los IRS I-II (38,39).
En el presente estudio las complicaciones agudas no fueron severas, la toxicidad hematológica leucopenia grado IV y trombocitopenia grado III solo se presentó en un paciente respectivamente, la mucositis y dermatitis grado III en solo un paciente respectivamente. En cuanto a las secuelas tardías solo se registró fibrosis grado II en un paciente con RMS en extremidad inferior. Existe probablemente un sub registro en las historias clínicas, porque muchos de estos pacientes no se controlaron a largo plazo en nuestros servicios, no se encontraron casos de neoplasias secundarias.
Referencias
1. Ries LAG, Harkins D, Krapcho M, Mariotto A, Miller BA, Feuer EJ, et al. SEER Cancer Statistics Review, 1975-2003, National Cancer Institute. Bethesda, MD. Disponible en: URL: http://seer.cancer.gov/csr/1975_2003.
2. Sankaran H, Danysh HE, Scheurer ME, Okcu MF, Skapek SX, Hawkins DS, et al. The role of childhood infections and immunizations on childhood rhabdomyosarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer. 2016;63:1557-1562.
3. Gripp KW, Scott CI Jr, Nicholson L, McDonald-McGinn DM, Ozeran JD, Jones MC, et al. Five additional Costello syndrome patients with rhabdomyosarcoma: Proposal for a tumor screening protocol. Am J Med Genet. 2002;108:80-87.
4. MacDonald S, Friedman A, Tarbell N. Rhabdomyosarcoma en: Halperin E, Constine L, Nancy T, editors. Pediatric Radiation Oncology. 5a edición. Filadelfia: Wolters Kluwer/Lippincott Williams & Wilkins. 2011.p.204.
5. Davis RJ, D'Cruz CM, Lovell MA, Biegel JA, Barr FG. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14)translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54:(11):2869-2872.
6. Kelly KM, Womer RB, Sorensen PH, Xiong QB, Barr FG. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol. 1997;15:1831-1836.
7. Scrable HJ, Witte DP, Lampkin BC, Cavenee WK. Chromosomal localization of the human rhabdomyosarcoma locus by mitotic recombination mapping. Nature. 1987;329:645-647.
8. Matthews D, Farewell V. Using and understanding medical statistics. 4a edición Nueva York: Karger;2007.
9. Roberts K, Ruiz F, Ruan L. Tumores pediátricos. En: Urdaneta N, Vera A, Peschel R, editores. Radioterapia Oncológica. 2ª edición. Caracas: Disinlimed;2009.p.1267.
10. Rockwell C. Principios de radiobiología. En: Urdaneta N, Vera A, Peschel R. Radioterapia Oncológica. 2ª edición. Caracas: Disinlimed;2009.p.86.
11. Raney RB, Walterhouse DO, Meza JL, Andrassy RJ, Breneman JC, Crist WM, et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: A report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J Clin Oncol. 2011;29:1312-1318.
12. Arndt CA, Stoner JA, Hawkins DS, Rodeberg DA, Hayes-Jordan AA, Paidas CN, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children's oncology group study D9803. J Clin Oncol. 2009;27:5182-5188.
13. Mercado GE, Barr FG. Fusions involving PAX and FOX genes in the molecular pathogenesis of alveolar rhabdomyosarcoma: Recent advances. Curr Mol Med. 2007;7:47-61.
14. Walterhouse DO, Pappo AS, Meza JL, Breneman JC, Hayes-Jordan AA, Parham DM, et al. Shorter-duration therapy using vincristine, dactinomycin, and lower-dose cyclophosphamide with or without radiotherapy for patients with newly diagnosed low-risk rhabdomyosarcoma: A report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J Clin Oncol. 2014;32:3547-3552.
15. Russell H, Swint JM, Lal L, Meza J, Walterhouse D, Hawkins DS, et al. Cost minimization analysis of two treatment regimens for low-risk rhabdomyosarcoma in children: A report from the Children's Oncology Group. Pediatr Blood Cancer. 2014;61:970-976.
16. Hawkins DS, Anderson JR, Mascarenhas L, Brian G, McCowage D, Rodeberg S, et al. Vincristine, dactinomycin, cyclophosphamide (VAC) vs. VAC/V plus irinotecan (VI) for intermediate-risk rhabdomyosarcoma (IRRMS): A report from the Children's Oncology Group Soft Tissue Sarcoma Committee. Disponible en: URL: http://meetinglibrary.asco.org/content/126983-144.
17. Weigel BJ, Lyden E, Anderson JR, Meyer WH, Parham DM, Rodeberg DA, et al. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: A report from the Children's Oncology Group. J Clin Oncol. 2016;34:117-122.
18. McMulkin HM, Yanchar NL, Fernandez CV, Giacomantonio C. Sentinel lymph node mapping and biopsy: A potentially valuable tool in the management of childhood extremity rhabdomyosarcoma. Pediatr Surg Int. 2003;19:453-456.
19. Neville HL, Andrassy RJ, Lally KP, Corpron C, Ross MI. Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg. 2000;35:961-964.
20. Andrassy RJ, Wiener ES, Raney RB, Lawrence W, Lobe TE, Corpron CA, et al. Thoracic sarcomas in children. Ann Surg. 1998;227:170-173.
21. Wiener ES, Anderson JR, Ojimba JI, Lobe TE, Paidas C, Andrassy RJ, et al. Controversies in the management of par testicular rhabdomyosarcoma: Is staging retroperitoneal lymph node dissection necessary for adolescents with resected paratesticular rhabdomyosarcoma? Semin Pediatr Surg. 2001;10:146-152.
22. Baker KS, Anderson JR, Link MP, Grier HE, Qualman SJ, Maurer HM, et al. Benefit of intensified therapy for patients with local or regional embryonal rhabdomyosarcoma: Results from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2000;18:2427-2434.
23. Wolden SL, La TH, LaQuaglia MP, Meyers PA, Kraus DH, Wexler LH. Long-term results of three-dimensional conformal radiation therapy for patients with rhabdomyosarcoma. Cancer. 2003;97:179-185.
24. Brand E, Berek JS, Nieberg RK, Hacker NF. Rhabdomyosarcoma of the uterine cervix. Sarcoma botryoides. Cancer. 1987;60:1552-1560.
25. Maurer HM, Beltangady M, Gehan EA, Crist W, Hammond D, Hays DM, et al. The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer. 1988;61:209-220.
26. Pappo AS, Lyden E, Breneman J, Wiener E, Teot L, Meza J, et al. Up-front window trial of topotecan in previously untreated children and adolescents with metastatic rhabdomyosarcoma: An intergroup rhabdomyosarcoma study. J Clin Oncol. 2001;19:213-219.
27. Ferrari A, Bisogno G, Casanova M, Meazza C, Piva L, Cecchetto G, et al. Paratesticular rhabdomyosarcoma: Report from the Italian and German Cooperative Group. J Clin Oncol. 2002;20:449-455.
28. Crist WM, Anderson JR, Meza JL, Fryer C, Raney RB, Ruymann FB, et al. Intergroup rhabdomyosarcoma study-IV: Results for patients with nonmetastatic disease. J Clin Oncol. 2001;19:3091-3102 .
29. Wharam MD, Hanfelt JJ, Tefft MC, Johnston J, Ensign LG, Breneman J, et al. Radiation therapy for rhabdomyosarcoma: Local failure risk for Clinical Group III patients on Intergroup Rhabdomyosarcoma Study II. Int J Radiat Oncol Biol Phys. 1997;38:797-804.
30. Pappo AS, Meza JL, Donaldson SS, Wharam MD, Wiener ES, Qualman SJ, et al. Treatment of localized nonorbital, nonparameningeal head and neck rhabdomyosarcoma: Lessons learned from intergroup rhabdomyosarcoma studies III and IV. J Clin Oncol. 2003;21:638-645.
31. Raney RB, Meza J, Anderson JR, Fryer CJ, Donaldson SS, Breneman JC, et al Treatment of children and adolescents with localized parameningeal sarcoma: Experience of the Intergroup Rhabdomyosarcoma Study Group protocols IRS-II through -IV, 1978-1997. Med Pediatr Oncol. 2002;38:22-32.
32. Wharam MD, Jr. Rhabdomyosarcoma of parameningeal sites. Semin Radiat Oncol. 1997;7: 212-216.
33. Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: A report from the children’s oncology group. J Clin Oncol.2002;20:2672-2679.
34. Donaldson SS, Meza J, Breneman JC, Crist WM, Laurie F, Qualman SJ, et al. Results from the IRS-IV randomized trial of hyper fractionated radiotherapy in children with rhabdomyosarcoma: A report from the IRSG. Int J Radiat Oncol Biol Phys. 2001;51:718-728.
35. Arcamone G, Giménez C, Pereira A, Reyes J, Gómez M, Mota D, et al. Rabdomiosarcoma en niños. Rev Venez Oncol. 2007;19(1):63-70.
36. Raney RB, Asmar L, Vassilopoulou-Sellin R, Klein MJ, Donaldson SS, Green J, et al. Late complications of therapy in 213 children with localized, nonorbital soft-tissue sarcoma of the head and neck: A descriptive report from the Intergroup Rhabdomyosarcoma Studies (IRS)-II and - III. IRS Group of the Children's Cancer Group and the Pediatric Oncology Group. Med Pediatr Oncol. 1999;33:362-371.
37. Raney B Jr, Heyn R, Hays DM, Tefft M, Newton WA Jr, Wharam M, et al. Sequelae of treatment in 109 patients followed for 5 to 15 years after diagnosis of sarcoma of the bladder and prostate. A report from the Intergroup Rhabdomyosarcoma Study Committee. Cancer. 1993;71:2387-2394.
38. Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE, Sanders SP. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;324:808-815.
39. Heyn R, Haeberlen V, Newton WA, Ragab AH, Raney RB, Tefft M, et al. Second malignant neoplasms in children treated for rhabdomyosarcoma. Intergroup Rhabdomyosarcoma Study Committee. J Clin Oncol. 1993;11:262-270.