SARCOMA DE EWING EXTRA ÓSEO DE ÚTERO
SARCOMA DE EWING EXTRA ÓSEO DE ÚTERO
Revista Venezolana de Oncología, vol. 32, núm. 3, pp. 175-180, 2020
Sociedad Venezolana de Oncología

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 4.0 Internacional.
Recepción: 25 Abril 2020
Revisado: 12 Mayo 2020
Aprobación: 10 Junio 2020
Resumen: El sarcoma de Ewing pertenece a un espectro de enfermedades neoplásicas conocidas como la familia de tumores de Ewing. La variante extra-ósea puede aparecer en tronco, extremidades y órganos del tracto genital femenino. OBJETIVO: Presentar el reporte de un caso de sarcoma de Ewing extra-óseo de útero. CASO CLÍNICO: Se trata de pacientes femenina de 48 años quien consultó por presentar sangrado vaginal, dolor y aumento de volumen abdominal de aproximadamente 6 meses de evolución. En la valoración del abdomen se palpó tumoración fija, firme, irregular y no dolorosa que se extendía hasta la cicatriz umbilical. El examen ginecológico demostró que la tumoración parecía ser de origen uterino, ocupando el fondo de saco de Douglas y separada anexos, con cuello uterino cerrado con sangrado de color oscuro en escasa cantidad. La ecografía abdomino-pélvica reveló tumoración que se extendía hacia el fondo de saco de Douglas. La paciente fue sometida a laparotomía encontrando tumoración bien definida y parcialmente encapsulada. Se realizó resección completa del tumor junto con histerectomía total, ooforosalpingectomía bilateral, linfadenectomía pélvica y para-aórtica, apendicetomía y omentectomía parcial. La evaluación histológica e histoquímica confirmaron el diagnóstico de sarcoma de Ewing extra-óseo del útero. CONCLUSIONES: El sarcoma de Ewing extra-óseo de útero es una entidad rara. Es importante identificar estos casos para mejorar las posibilidades de supervivencia. La sintomatología es inespecífica y similar a otras patologías pélvicas extra-intestinales. La histología e inmunohistoquímica son fundamentales para el diagnóstico definitivo y tratamiento oportuno.
Palabras clave: Sarcoma de Ewing, útero, extra-óseo, tumor neuroectodérmico.
Abstract: The Ewing's sarcoma belongs to a spectrum of neoplastic diseases known as Ewing's family of tumors. The extra-osseous variant may appear in trunk, extremities, and organs of the female genital tract. OBJECTIVE: We present a case report of extra-osseous Ewing's sarcoma of the uterus. CLINICAL CASE: A 48-year old female patient who consulted for presenting vaginal bleeding, pain and abdominal volume increase of approximately 6 months of evolution. In the evaluation of the abdomen, a fixed, firm, irregular and painless tumor was found that extended to the umbilical scar. Gynecological examination showed that the tumor appeared to be of uterine origin, occupying the bottom of the sac of Douglas and separated from both adnexa, with a closed cervix with dark bleeding in a small amount. The abdominal pelvic ultrasound revealed a tumor that extended towards the bottom of the sac of Douglas. The patient underwent laparotomy, finding a well-defined and partially encapsulated tumor. The complete tumor resection was performed along with a total hysterectomy, bilateral oophorosalpingectomy, pelvic and para-aortic lymphadenectomy, appendectomy, and the partial omentectomy. The histological and the histochemical evaluation confirmed the diagnosis of extra-osseous Ewing's sarcoma of the uterus. CONCLUSIONS: The Ewing’s sarcoma of the uterus is a rare entity. It is important to identify these cases to improve chances of survival. The symptomatology is nonspecific and similar to other extra intestinal pelvic pathologies. The histology and the immunohistochemistry are fundamental for definitive diagnosis and the opportune treatment.
Keywords: Ewing's sarcoma, uterus, extra-osseous, neuroectodermal tumor.
INTRODUCCIÓN
El sarcoma de Ewing y el tumor neuro-ectodérmico periférico son una única entidad que posee diversos grados de diferenciación neuro-ectodérmica. Surgen de células progenitoras mesenquimales, formando un espectro de enfermedades neoplásicas, conocido como familia de tumores de Ewing (1). Estos tienen como características la translocación recíproca entre los cromosomas 11 y 22 [t (11; 22) (q24; Q12)] que forman el producto de fusión EWSR1-FLI1, así como características histopatológicas e inmunohistoquímicas similares (1,2). La familia consta de los siguientes cuatro subtipos: sarcoma óseo de Ewing, sarcoma de Ewing extra-óseo, tumor neuro-ectodérmico primitivo periférico y tumor de Askin (2).
El sarcoma de Ewing y el tumor neuro-ectodérmico periférico son una única entidad que posee diversos grados de diferenciación neuro-ectodérmica. Surgen de células progenitoras mesenquimales, formando un espectro de enfermedades neoplásicas, conocido como familia de tumores de Ewing (1). Estos tienen como características la translocación recíproca entre los cromosomas 11 y 22 [t (11; 22) (q24; Q12)] que forman el producto de fusión EWSR1-FLI1, así como características histopatológicas e inmunohistoquímicas similares(1,2). La familia consta de los siguientes cuatro subtipos: sarcoma óseo de Ewing, sarcoma de Ewing extra-óseo, tumor neuro-ectodérmico primitivo periférico y tumor de Askin (2).
El sarcoma de Ewing tiene una incidencia anual de menos de 1 caso por millón con incidencia máxima a los 10 - 20 años de edad, es poco frecuente en adultos mayores de 30 años y la mayor frecuencia de casos se produce en el sexo masculino (relación de 5:1)(3,4). Los sarcomas de Ewing extra-óseos son infrecuentes. Existen informes de casos y series que indican apariciones inusuales en cavidad oral, glándulas salivales, pulmón, corazón, pericardio, esófago, estómago, páncreas, próstata, riñón, vejiga urinaria, útero, ovarios y vagina (5). Los tumores del tracto genital femenino son raros pero devastadores, siendo el sitio más común de aparición el ovario. Los sarcomas de origen uterino y cervical son difíciles de diferenciar de los leiomiomas, lo que lleva a posibles retrasos diagnósticos y terapéuticos (3). Se presenta un caso de sarcoma de Ewing extra-óseo de útero.
CASO CLÍNICO
Se trata de paciente femenina de 48 años, X gestas, VIII paras, II abortos, quien consultó por presentar sangrado vaginal acompañado de dolor abdominal de leve intensidad y aumento de volumen abdominal de aproximadamente 6 meses de evolución. La paciente refería pérdida de peso y del apetito. Negaba antecedentes médicos o quirúrgicos de importancia.
En la valoración del abdomen se palpó tumoración fija, firme, irregular y no dolorosa que se extendía hasta la cicatriz umbilical. No se palparon visceromegalias ni adenopatías. El examen ginecológico demostró que la tumoración parecía ser de origen uterino, ocupando el fondo de saco de Douglas y separada de anexos, con cuello uterino cerrado con sangrado de color oscuro en escasa cantidad. De igual forma, el tacto rectal demostró que la tumoración se ubicaba anterior al recto (lesión extra-luminal) sin afectación de la mucosa. El resto del examen físico y los signos vitales estaba dentro de límites normales. Los exámenes de laboratorio reportaron valores de hemoglobina de 8,1 g/L y hematocrito de 27 %. No se encontraron alteraciones en el resto de los resultados de laboratorio.
La ecografía abdomino-pélvica reveló que la tumoración medía 20 cm x 13 cm x 9 cm encontrándose una lesión en cara posterior que medía 6 cm x 4 cm x 3 cm que se extendía hacia el fondo de saco de Douglas. Los anexos estaban desplazados hacia delante con pequeña cantidad de líquido libre en cavidad abdominal. El hígado bazo y páncreas estaban normales. No se observaron evidencias de carcinomatosis y ambos anexos estaban normales. La ecografía Doppler confirmó la naturaleza hiper-vascular del tumor. Las imágenes de tomografía computada demostraron la presencia del tumor de origen pélvico, que desplazaba asas intestinales (Figura 1). No se encontró ascitis, carcinomatosis y adenopatías retroperitoneales. Los valores de los marcadores tumorales fueron CA125: 91,5 UI/mL, alfafetoproteína: 315 ng/mL, y deshidrogenasa láctica 1 059 UI/mL. Los valores de gonadotropina coriónica y antígeno carcino-embriogénico estaban dentro de límites normales. Tanto la endoscopia superior como la colonoscopia no mostraron alteraciones. La radiografía simple de tórax estaba normal.
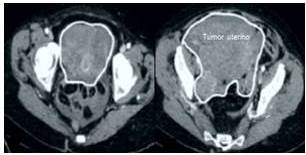
Figura 1
Tomografía computada que demuestra tumoración pélvica que desplazaba las asas intestinales sin signos de metástasis visceral
La paciente fue sometida a laparotomía exploratoria encontrando una tumoración bien definida y parcialmente encapsulada de 21 cm x 14 cm x 11 cm que se originaba desde el útero y ocupaba la pelvis desplazando recto, colon sigmoides y ascendente acompañada de escaso líquido, el cual fue recolectado y enviado a evaluación citológica. Los anexos estaban normales sin lesiones macroscópicas evidentes. No se encontró evidencia de metástasis hepáticas ni aumento de tamaño de ganglios linfáticos locales. La vesícula biliar, estómago, páncreas y apéndice cecal estaban normales. La biopsia por congelación del tumor sugirió la posibilidad diagnóstica de sarcoma de Ewing / tumor neuro-ectodérmico primitivo periférico por lo que se realizó resección completa del tumor junto con histerectomía total, ooforosalpingectomía bilateral, linfadenectomía pélvica y para-aórtica, apendicetomía y omentectomía parcial.
La evaluación macroscópica reveló tumor parcialmente encapsulado (Figura 2). Los cortes de la pieza mostraron áreas de necrosis y hemorragia. Al examen microscópico demostró láminas de células redondas con núcleos grandes redondos-ovalados y citoplasma claro las cuales eran positivas a la técnica de coloración de Schiff. Las zonas de necrosis mostraron proliferación periteliomatosa focal de células tumorales alrededor de los vasos sanguíneos con mitosis incrementada. La coloración inmunohistoquímica mostró co-expresión de CD99, vimentina y S-100. También demostró que las células eran débilmente positivas a enolasa neuroespecífica y CD56. Tanto la reactividad a pancitoqueratina, desmina, como a sinaptofisina, CK20, CD45 y cromogranina fue negativa (Figura 3). La citología del líquido peritoneal y las secciones del epiplón, ganglios linfáticos, endometrio, trompas de Falopio, cuello uterino y ovarios estaban normales sin evidencia de alteraciones malignas. El estudio de la hibridación de fluorescencia in situ varias semanas después reveló reordenamiento del gen ESWR1 en 90 % de las células, lo que confirmó el diagnóstico de sarcoma de Ewing extra-óseo del útero.
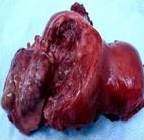
Figura 2.
Apariencia macroscópica del sarcoma de Ewing extra-óseo.
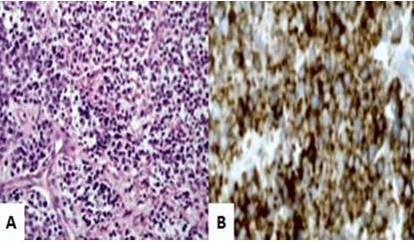
Figura 3
Imágenes histopatológicas de la tumoración que demuestra A) Laminas de células redondas y pequeñas uniformes con patrón trabecular. Coloración hematoxilina-eosina. 100X B) Inmunotinción para CD 99 difusa. 400X.
La paciente cursó el período posoperatorio sin complicaciones y fue referida al servicio de oncología de otro hospital en el cual indicaron la evaluación tomográfica de tórax, abdomen y pelvis, las cuales fueron negativas. La gammagrafía ósea y biopsia de médula ósea resultaron negativas para la metástasis. Se inició tratamiento con quimioterapia planificado por oncología médica. Ocho meses después del tratamiento, la paciente continuaba en seguimiento ambulatorio sin evidencia de recurrencia.
DISCUSIÓN
El sarcoma de Ewing extra-óseo de útero es una entidad extremadamente rara. Este informe de caso demuestra la importancia de considerar esta entidad dentro de los diagnósticos diferenciales de los tumores uterinos. A diferencia del sarcoma óseo de Ewing, que es más frecuente en el sexo masculino, no existe diferencia de géneros en la frecuencia de los sarcomas de Ewing extra-óseos y más del 70 % de los casos son diagnosticados en la adolescencia(5,6,7). Existen aproximadamente 80 casos reportados, de los cuales 60 fueron en cuerpo uterino y 21 casos en cuello uterino (8).
Las pacientes con sarcoma de Ewing presentan con frecuencia retraso en la aparición de la sintomatología y el diagnóstico temprano es bastante raro, en especial los que aparecen en la pelvis, porque no son palpables hasta que alcanzan grandes dimensiones (3). Casi todos los casos tienen enfermedad diseminada oculta al momento del diagnóstico (5,6,7). Los síntomas más frecuentes son sangrado genital irregular (85 % en los casos de cuello uterino y 70 % en tumores del cuerpo uterino) y dolor pélvico o lumbar (27 % de los casos), el cual puede irradiarse a las extremidades inferiores. Otros signos / síntomas que pueden aparecer incluyen flujo vaginal, disuria, polaquiuria fiebre y pérdida de peso (8). Los signos neurológicos, como compresión de las raíces nerviosas, están presentes en cerca de la mitad de los casos (2). Entre los hallazgos físicos más comunes se observan lesiones nodulares que se extienden hacia la pared vaginal y útero aumentado de tamaño, lo que puede llevar a confundirlo con leiomiomas uterinos (8).
El sarcoma de Ewing puede aparecer en región paravertebral, área retroperitoneal, pared torácica y extremidades inferiores. No existen características radiológicas especificas; sin embargo, la mayoría de las lesiones están encapsuladas y son hipoecoicas en la ecografía convencional, hipodensas en la tomografía computada e iso-hipointensas en T1 e hiperintensas en T2 en las imágenes de resonancia magnética (6). Estas últimas también son el método ideal para evaluar la afección de los tejidos blandos circundantes (2,6). La tomografía por emisión de positrones puede ser útil en el diagnóstico, estadificación y seguimiento de la respuesta al tratamiento (8).
El diagnóstico es difícil por microscopía de rutina, porque su morfología es similar a otros tumores. En la mayoría de los casos se observan láminas de células azules pequeñas con cromatina nuclear punteada y ausencia de nucléolos. La necrosis de células adyacentes es común. Este tipo de células pequeñas células azules redondas en neoplasias también está presente en linfoma linfoblástico, meduloblastoma, tumor neuroendocrino de células pequeñas y neuroblastoma. Otros sarcomas de tejidos blandos y el rabdomiosarcoma también deben considerarse en el diagnóstico diferencial (9).
La inmunohistoquímica es crítica para el diagnóstico, porque los carcinomas neuroendocrinos de células pequeñas suelen ser positivos para cromogranina, sinaptofisina y enolasa específica de las neuronas. La gran mayoría de los sarcomas de Ewing expresan la glicoproteína de superficie celular CD99 o antígeno de superficie MIC2 que es codificado por el gen CD99 (10). La expresión de MIC2 por un tumor maligno de células azules pequeñas es un marcador de diagnóstico sensible para la familia de tumores de Ewing. El MIC2 carece de especificidad porque otros tumores, como el rabdomiosarcoma, pueden expresarlo. Sin embargo, los carcinomas de células pequeñas son negativos. También se utilizan marcadores linfoideos, antígeno común de leucocitos, CD20 y CD3 para diferenciarlo del linfoma (11).
La caracterización genética molecular de anomalías cromosómicas específicas de la familia de tumores de Ewing ha mejorado la detección. Las translocaciones características involucran el gen EWSR1 en 22q12.2 y varios otros genes específicos de la familia de transcripción de eritroblastos en 11q24.1-q24.3 y que se observa en más de 90 % de los casos (5,12). El ESWR codifica una proteína multifuncional que regula varios procesos celulares. El FLI1 codifica una proteína que controla tanto el desarrollo como la proliferación celular y carcinogénesis (13). La translocación EWSR1-ERG [t (21; 22) (q22; q12)] está presente en 5 %-10 % de los casos, mientras que otras translocaciones son menos comunes (13,14).
Actualmente no existe uniformidad en el tratamiento, principalmente debido a la baja frecuencia de esta neoplasia. No obstante, el control de la enfermedad con cirugía, quimioterapia con múltiples agentes y radioterapia son pilares terapéuticos en estos casos, porque la mayoría tienen metástasis ocultas. Las combinaciones del tratamiento deben considerar: edad, localización anatómica, estadio de la enfermedad, tamaño del tumor, presencia de metástasis y potencial respuesta al tratamiento. La opción quirúrgica recomendada en los tumores del tracto genital es la histerectomía abdominal radical y ooforosalpingectomía bilateral con o sin disección de ganglios linfáticos pélvicos en aquellos casos en estadios tempranos. En aquellas pacientes en los que no se pueda realizar se recomienda la resección amplia del tumor (8,10).
El sarcoma de Ewing es una neoplasia maligna sensible a la quimioterapia, la cual permite controlar la enfermedad y aumentar la tasa de supervivencia a largo plazo. Datos previos demuestran una tasa de supervivencia de 5 años de 75 % en aquellos casos con tumores no metastásicos tratados con resección quirúrgica y quimioterapia comparados con 25 % en pacientes que no se realizó la cirugía (3,4). La quimioterapia con múltiples agentes ha demostrado que aumenta la tasa de supervivencia de 10 % a 70 % (3). Los agentes quimioterapéuticos más utilizados incluyen doxorubicina, ciclofosfamida, vincristina, actinomicina D, ifosfamida y etopósido y la combinación más evaluada es vincristina, doxorubicina y ciclofosfamida (régimen de elección para el sarcoma óseo de Ewing). La ifosfamida es uno de los agentes quimioterapéuticos más eficaces en el tratamiento de estos tumores, en especial en aquellos casos con tumores resistentes a otros fármacos (15). La combinación de fármacos puede lograr una supervivencia a largo plazo del 70 % - 80 % de los casos con enfermedad no metastásica (3). De igual forma, la radioterapia también puede ser útil en el tratamiento de estos pacientes, particularmente en aquellos con enfermedad residual después de la cirugía y quimioterapia (2).
El sarcoma de Ewing extra-óseo de útero es una entidad rara que plantea diferentes dilemas clínicos. Es importante identificar estos casos para mejorar las posibilidades de supervivencia con las modalidades terapéuticas disponibles en la actualidad. Desafortunadamente la sintomatología es inespecífica y similar a otras patologías pélvicas extra-intestinales. La histología e inmunohistoquímica son fundamentales para el diagnóstico definitivo y tratamiento oportuno. La detección de más casos en el futuro ayudara a establecer patrones de tratamiento y seguimiento.
REFERENCIAS
1. Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, Sorna V, et al. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res. 2014;20(17):4584-4597.
2. Saif MW, Kaley K. Extra-osseous Ewing's sarcoma of the pancreas: An uncommon but treatable disease. Cureus. 2017;9(11):e1882.
3. Van Mater D, Wagner L. Management of recurrent Ewing sarcoma: Challenges and approaches. Onco Targets Ther. 2019;12:2279-2288.
4. Suarez Antelo J, Rodriguez Garcia C, Montero Martinez C, Verea Hernando H. Pulmonary Ewing sarcoma/primitive neuroectodermal tumor: A case report and a review of the literature. Arch Bronconeumol. 2010;46(1):44-46.
5. Aurias A, Rimbaut C, Buffe D, Zucker JM, Mazabraud A. Translocation involving chromosome 22 in Ewing's sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet Cytogenet. 1984;12(1):21-25.
6. Khosla D, Verma S, Punia RS, Dass A, Dimri K, Kaur G, et al. Extra-osseous Ewing's sarcoma of the parapharyngeal space- a rare entity - with review of literature. Iran J Otorhinolaryngol. 2019;31(102):51-54.
7. De Nola R, Di Naro E, Schonauer LM, Lucarelli G, Battaglia M, Fiore MG, et al. Clinical management of a unique case of PNET of the uterus during pregnancy, and review of the literature. Medicine (Baltimore). 2018;97(2):e9505.
8. Mashriqi N, Gujjarlapudi JK, Sidhu J, Zur M, Yalamanchili M. Ewing's sarcoma of the cervix, a diagnostic dilemma: A case report and review of the literature. J Med Case Rep. 2015;9:255.
9. Wang X, Gao Y, Xu Y, Liu Y, Qu P. Primary primitive neuroectodermal tumor of the cervix: A report of two cases and review of the literature. Mol Clin Oncol. 2017;6(5):697-700.
10. Kilpatrick SE, Reith JD, Rubin B. Ewing sarcoma and the history of similar and possibly related small round cell tumors: From whence have we come and where are we Going? Adv Anat Pathol. 2018;25(5):314-326.
11. Khosla D, Rai B, Patel FD, Sreedharanunni S, Dey P, Sharma SC. Primitive neuroectodermal tumor of the uterine cervix diagnosed during pregnancy: A rare case with review of literature. J Obstet Gynaecol Res. 2014;40(3):878-882.
12. Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, et al. A variant Ewing's sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene. 1995;10(6):1229-1234.
13. Arvand A, Denny CT. Biology of EWS/ETS fusions in Ewing's family tumors. Oncogene. 2001;20(40):5747-5754.
14. Stegmaier S, Leuschner I, Aakcha-Rudel E, Münch P, Kazanowska B, Bekassy A, et al. Identification of various exon combinations of the ews/fli1 translocation: An optimized RT-PCR method for paraffin embedded tissue -- a report by the CWS-study group. Klin Padiatr. 2004;216(6):315-322.
15. Akazawa M, Saito T, Ariyoshi K, Okadome M, Yokoyama R, Taguchi K. Adjuvant chemotherapy for a primitive neuroectodermal tumor of the uterine corpus: A case report and literature review. J Obstet Gynaecol Res. 2018;44(10):2008-2015.
Notas de autor
sippenbauch@gmail.com