Caso clínico de interés especial
Síndrome de fibromatosis hialina: reporte de un caso y revisión bibliográfica
Hyaline fibromatosis syndrome: A case report and literature review.
Síndrome de fibromatosis hialina: reporte de un caso y revisión bibliográfica
Acta Pediátrica de México, vol. 40, núm. 5, pp. 274-281, 2019
Instituto Nacional de Pediatría

Esta obra está bajo una Licencia Creative Commons Atribución 4.0 Internacional.
Recepción: 28 Octubre 2018
Aprobación: 12 Agosto 2019
Resumen:
ANTECEDENTES: El síndrome de fibromatosis hialina es una enfermedad rara del tejido conectivo, con patrón de herencia autosómica recesiva, caracterizada por múltiples nódulos subcutáneos, en la piel, hipertrofia gingival, contracturas articulares, entre otras. CASO CLÍNICO: Paciente de 5 años, con antecedentes familiares de un hermano fallecido a los 2 años por bronconeumonía, con contracturas articulares desde el nacimiento, desviación cubital de ambos brazos y luxación de cadera, diagnosticado con artrogriposis múltiple congénita. El caso aquí expuesto, desde el nacimiento padeció contracturas, falta de extensión y flexión en los miembros superiores e inferiores, y desviación cubital en ambas manos. El primer año de vida manifestó lesiones papulares en el cuello y la región perianal, y lesiones nodulares subcutáneas en ambos pabellones auriculares, hipertrofia gingival, hiperpigmentación en los sitios de presión y protrusiones óseas a partir de los 2.5 años. A los 3 años acudió al Centro de Rehabilitación e Inclusión Teletón (CRIT) Estado de México, con diagnóstico de artrogriposis múltiple congénita. Las radiografías de las extremidades superiores e inferiores evidenciaron osteopenia generalizada y aumento de la radiolucencia en forma difusa. La biopsia de los nódulos auriculares subcutáneos reportó material hialino PAS positivo. Con base en los hallazgos clínicos se estableció el diagnóstico de síndrome de fibromatosis hialina. El tratamiento consistió en terapias físicas, ocupacionales, pulmonares y de lenguaje, con reacción favorable. Sus condiciones físicas generales empeoraron a partir de los 5 años; inició con artralgias severas, infecciones pulmonares recurrentes y diarrea de difícil control. Falleció a los 8 años por bronconeumonía adquirida en la comunidad. CONCLUSIONES: El diagnóstico inicial del síndrome de fibromatosis hialina se establece por los hallazgos clínicos. Es importante establecer el diagnóstico diferencial con artrogriposis múltiple congénita.
Palabras clave: Síndrome de fibromatosis hialina, hipertrofia gingival, bronconeumonía, artrogriposis múltiple congénita.
Abstract:
BACKGROUND: Hyaline fibromatosis syndrome is an exceptional disease of the connective tissue, with an autosomal recessive inheritance pattern, characterized by multiple subcutaneous nodules in the skin, gingival hypertrophy, joint contractures, among others. CLINICAL CASE: 5-year-old pediatric patient, with a family history of a brother who died at 2 years of age due to bronchopneumonia, with joint contractures from birth, ulnar deviation of both arms and hip dislocation, diagnosed with congenital multiple arthro- gryposis. The case presented here, from birth suffered contractures, lack of extension and flexion in the upper and lower limbs, and ulnar deviation in both hands. The first year of life he showed papular lesions in the neck and perianal region, and subcutaneous nodular lesions in both auricular folds gingival hypertrophy, hyperpigmentation at the pressure sites and bone protrusions from 2.5 years. At 3 years he went to the Centro de Rehabilitación e Inclusión Teletón (CRIT) Estado de México, with a diagnosis of congenital multiple arthrogryposis. Radiographs of the upper and lower extremities showed widespread osteopenia and increased radiolucence diffusely. Subcutaneous atrial nodule biopsy reported PAS positive hyaline material. Based on the clinical findings, the diagnosis of hyaline fibromatosis syndrome was established. The treatment consisted of physical, occupational, pulmonary and language therapies, with a favorable outcome. His general physical conditions worsened after 5 years; he began with severe arthralgia recurrent lung infections and diarrhea that was difficult to control. He died at age 8 from bronchopneumonia acquired in the community. CONCLUSIONS: The initial diagnosis of patients with hyaline fibromatosis syndrome is established by clinical findings. It is important to establish the differential diagnosis with congenital multiple arthrogryposis.
Keywords: Hyaline fibromatosis syndrome, Gingival hypertrpphy, Bronchopneumonia, Congenital multiple artrogryposis.
ANTECEDENTES
El síndrome de fibromatosis hialina es una enfermedad autosómica recesiva (MIM #228600), panétnica, poco frecuente, descrita por primera vez en 1873, como una variante de la neurofibromatosis, denominada molusco fibroso. Se caracteriza por la aparición de nódulos subcutáneos, fibrosis hialinizante de la piel y los órganos en forma sistémica, hiperplasia gingival, contracturas articulares y lesiones osteolíticas en los huesos largos, que suele manifestarse durante la infancia temprana. Hasta la fecha se han reportado 169 casos.1,2,3,4,5
El síndrome de fibromatosis hialina se origina por variantes patogénicas en el gen que codifica para el receptor 2 de la toxina del ántrax (ANTX2), también conocido como proteína capilar morfogénica-2 (CMG2), con locus en 4q21.
El gen ANTXR2/CMG2 codifica para una proteína que contiene un péptido de señal, un dominio tipo A extracelular del factor de von Willebrand (vWA), un dominio similar a la inmunoglubulina, un dominio transmembrana y una porción final citosólica.6,7
Las mutaciones en el gen ANTXR2 alteran la síntesis de glucosaminoglicanos por los fibroblastos, lo que provoca deficiencia del metabolismo del colágeno. Se han propuesto tres posibles vías por las que se altera la permeabilidad de la membrana basal de los tejidos, provocando las lesiones papulares y nodulares típicas de la enfermedad:1) aumento en la síntesis y degradación del colágeno tipo I, 2) reducción significativa del metabolismo del colágeno tipo III y IV y 3)depósitos de colágeno tipo VI.1,2,4,8,9,10,11 La pérdida de la función de CGM2 provoca la acumulaciónde colágeno VI en la matriz extracelular, con subsiguiente ruptura del tejido a largo plazo.6
Las manifestaciones iniciales aparecen en los primeros meses de vida,6,7 con contracturas articulares y lesiones pápulo-nodulares aperladas en el cuello y la región periorificial (oral, nasal y anal), acompañadas de hipertrofia gingival progresiva.12,13
El diagnóstico de síndrome de fibromatosis hialina se establece mediante hallazgos clínicos, con apoyo del examen histopatológico de tejido cutáneo o intestinal, en el que se identifica la acumulación de material hialino14,15 y se confirma con el análisis molecular del gen ANTXR2 (único gen identificado, hasta hoy, asociado con la enfermedad).2,4,15 Se han descrito cerca de 50 variantes patogénicas del gen y en aproximadamente 80% de los casos los genotipos son homocigotos. Los “puntos calientes de mutación o sitios recurrentes mutados (hotspot)” se encuentran en el dominio vWA del exón 13, donde c.1073dup y c.1074del son las mutaciones más frecuentes.7
CASO CLÍNICO
Niño de 5 años, mexicano, nacido del cuarto embarazo de una madre de 35 y padre de 43 años, no consanguíneos, originarios de una comunidad posiblemente endogámica de Teziutlán, Puebla (México), aparentemente sanos. Antecedentes maternos de un aborto espontáneo y familiares de un hermano sano y otro finado a los 2 años, por bronconeumonía con contracturas articulares desde el nacimiento, con desviación cubital de ambos brazos y luxación de caderas, diagnosticado clínicamente en su comunidad de origen con artrogriposis múltiple congénita, con mutación de novo.
La gestación transcurrió normal, de término, el nacimiento fue por cesárea, por antecedente de doble circular de cordón umbilical al cuello y sufrimiento fetal. Al nacimiento registró: peso de 3160 g, talla de 52 cm, Apgar 8/9, sin cianosis ni datos de asfixia. Desarrollo psicomotor con lenguaje acorde con la edad y retraso en los hitos del desarrollo motor por la alteración de base.
Desde el nacimiento se observaron contracturas, con falta de extensión y flexión en los miembros superiores e inferiores, y en ambas manos se observaba desviación cubital. Debido al antecedente de un hermano fallecido por artrogriposis múltiple congénita, se consideró que cumplía con los criterios clínicos de la enfermedad, con patrón de herencia autosómica recesiva y antecedentes familiares de endogamia.
Desde el primer año de vida manifestó lesiones papulares aperladas en la cara posterior del cuello y la región perianal, además de lesiones nodulares subcutáneas en ambos pliegues auriculares, hipertrofia gingival e hiperpigmentación en los sitios de presión y protrusiones óseas a partir de los 2 años y medio de edad. Figuras 1 y 2
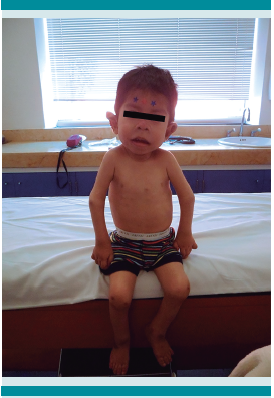
Figura 1.
Fenotipo del paciente: nariz bulbosa, discretamente tosca, pabellones auriculares prominentes, hipotrofia muscular generalizada, contracturas en grandes articulaciones, manos en garra.
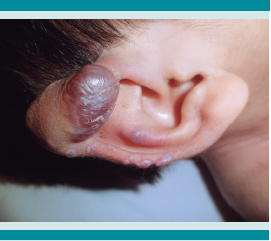
Figura 2.
Nódulos de coloración violácea en hélix en los pabellones auriculares, típicos del síndrome de fibromatosis hialina.
A la exploración física se encontraron: talla baja, facies tosca, nódulo en la región temporal derecha, cejas escasas, nariz ancha con narinas antevertidas, hipertrofia gingival importante; pabellones auriculares bien implantados, con múltiples nódulos de coloración violácea en el pliegue; cuello con lesiones aperladas y piel gruesa (Figura 2). Tórax simétrico, discretamente excavatum, abdomen globoso sin megalias y ano con lesiones nodulares. Las extremidades superiores e inferiores permanecían íntegras, hipotróficas, con limitación a la extensión y flexión en los codos, las rodillas y caderas; manos en garra, control volitivo disminuido para su edad, con predominio del patrón flexor y percepción de dolor durante la movilización de las extremidades. A los 3 años, la familia acudió al CRIT Estado de México para recibir atención en forma integral, enviados de su clínica familiar con diagnóstico de artrogriposis múltiple congénita.
El cariotipo en sangre periférica, bandas GTG, no reportó alteraciones (46, XY [20], 550 bandas). Las radiografías de las extremidades superiores e inferiores evidenciaron osteopenia generalizada y aumento de la radiolucencia en forma difusa. La biopsia de nódulos auriculares subcutáneos reportó material hialino PAS positivo. Con base en los hallazgos clínicos se estableció el diagnóstico de síndrome de fibromatosis hialina.
El paciente recibió tratamiento multi e interdisciplinario, con terapias físicas, ocupacionales, pulmonares, de lenguaje y tanque terapéutico. La familia recibió atención integral psicológica y tanatológica, con la finalidad de estimular el desarrollo del paciente (según sus capacidades residuales), mejorar su calidad de vida, lograr la aceptación de los padres de la discapacidad, comprender el pronóstico y evolución del caso, y recibir el asesoramiento genético certero.
Durante su estancia hospitalaria tuvo evolución poco satisfactoria; se observó disminución de los arcos de movilidad al flexionar los hombros y la cadera a 90º. Logró controlar el cuello y el tronco; toleró la sedestación con apoyo, sin dolor.
A los 4 años manifestó escoliosis dorsolumbar dextrocóncava de 34° y torácica de 18°; mejoró su lenguaje y tuvo adecuada comprensión pragmática para su edad, con disglosias secundarias a la hiperplasia gingival, propia del síndrome. Inició el proceso de lectoescritura mediante asistencia tecnológica, consolidándose a los 4 años y 5 meses (con apoyo de una tableta electrónica) e integrándose al nivel preescolar regular.
Su condición física general empeoró a partir de los 5 años; inició con artralgias severas, incluso en reposo, infecciones pulmonares recurrentes y diarrea de difícil control, con subsiguiente desnutrición en forma crónica. Falleció a los 8 años por bronconeumonía adquirida en la comunidad.
No se efectuó la identificación de las variantes del gen ANTXR2, debido a las condiciones generales del paciente y porque la familia no pudo solventar el costo del estudio molecular. Se decidió asesorar a los padres acerca de la enfermedad, por considerar que ambos eran portadores sanos de la alteración y descartar el diagnóstico de artrogriposis múltiple congénita, establecido al nacimiento del paciente.
DISCUSIÓN
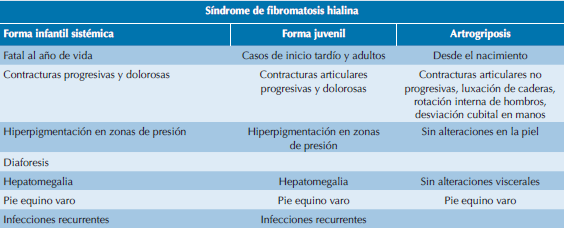
El síndrome de fibromatosis hialina suele manifestarse durante la infancia temprana y el pronóstico a largo plazo es malo. Algunos autores proponen que la hialinosis sistémica infantil es la forma más grave de la enfermedad (incluso provoca la muerte), mientras que la fibromatosis hialina juvenil representa la forma leve.3,10,16 Diversos estudios demuestran que ambas enfermedades se originan por mutaciones del gen ANTXR2.2,4 Es una alteración alélica, caracterizada por depósitos hialinos perivasculares.17,18,19 El Cuadro 1 muestra la comparación del síndrome de fibromatosis hialina y artrogriposis múltiple congénita.
Hanks y sus colaboradores17 reportaron la correlación genotipo-fenotipo en 17 familias con la manifestación sistémica infantil y juvenil de la enfermedad, y observaron que los pacientes con lectura o variantes de splicing solían tener un fenotipo compatible con la forma infantil sistémica; mientras que las variantes sin sentido o de sentido equivocado en el dominio citoplasmático se asociaron con el fenotipo de la enfermedad juvenil leve, con supervivencia hasta la etapa adulta, sin padecer infecciones recurrentes, diarrea ni insuficiencia multiorgánica. En todos los casos con confirmación molecular de alguna de ambas variantes clínicas se informaron manifestaciones esqueléticas similares a la artrogriposis múltiple congénita, con contracturas articulares en los codos, las muñecas, rodillas y caderas.17
El diagnóstico diferencial del síndrome de fibromatosis hialina se establece con artrogriposis múltiple congénita (contracturas articulares en los codos y las rodillas, hemangiomas faciales, rotación interna en ambos miembros superiores y luxación de caderas),20 síndrome de Winchester (facies tosca, hipertricosis, piel gruesa e hiperpigmentada y opacidad corneal),19 síndrome de nodulosis-artropatía-osteolisis (de curso clínico grave, múltiples nódulos dolorosos subcutáneos, osteoporosis generalizada, osteolisis masiva en las manos y los pies) y síndrome de Torg (múltiples nódulos subcutáneos no dolorosos asociados con osteoporosis y osteolisis en las manos y los pies). Estas enfermedades son de origen alélico, autosómicas recesivas y provocadas por mutaciones del gen MMP2 (matriz metaloproteinasa-2), con proliferación de fibroblastos y engrosamiento de colágeno.1,2También deben considerarse en el diagnóstico diferencial enfermedades de depósito lisosomal, que suelen manifestarse por disostosis múltiple, mucolipidosis II (enfermedad de células-I) y polidistrofia pseudo-Hurler (mucolipidosis III-A).15, 19
El caso aquí expuesto cumplió con los criterios clínicos del síndrome de fibromatosis hialina variante infantil, por la manifestación de nódulos subcutáneos auriculares, lesiones nodulares aperladas en el cuello, hipertrofia gingival, contracturas articulares con hiperpigmentación en los sitios de presión, dolor articular grave, osteopenia, desnutrición, infecciones recurrentes respiratorias, diarrea crónica y, finalmente, fallecimiento en forma temprana, confirmado con el estudio histológico de depósitos hialinos PAS positivos. El Cuadro 2 muestra la comparación de los casos clínicos descritos en la bibliografía con síndrome de fibromatosis hialina infantil, incluido el nuestro, con los principales datos clínicos de los pacientes.21,22,23,24,25,26,27
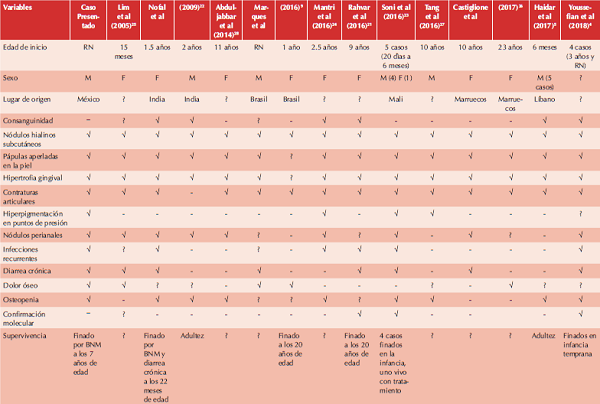
Recién nacido (RN); masculino (M); Femenino (F); no reportado (?); presente (√); ausente (-).
La movilidad articular en pacientes con síndrome de fibromatosis hialina suele mejorar con la administración de penicilamina,1,2 antiinflamatorios no esteroides, opiáceos y gabapentina para el tratamiento del dolor, además de fisioterapia. Algunos casos requieren tratamiento paliativo.
En pacientes con síndrome de malabsorción y linfangiectasia debe considerarse la colocación de una sonda nasogástrica para asegurar la alimentación adecuada; las infecciones deben tratarse de acuerdo con el sitio de infección y el agente causal; en caso de lesiones que obstruyan la vía aérea, éstas deberán intervenirse quirúrgicamente, aunque el riesgo de recurrencia es alto.15
CONCLUSIONES
El diagnóstico inicial de pacientes con síndrome de fibromatosis hialina se establece mediante hallazgos clínicos, logrando diferenciar entre los subtipos infantil y juvenil de la enfermedad, según la evolución del cuadro, supervivencia y, sobre todo, tomando en cuenta el diagnóstico diferencial de artrogriposis múltiple congénita. En las familias de bajos recursos, que no pue- den solventar el gasto del estudio molecular confirmatorio, el diagnóstico clínico puede ser suficiente para ofrecer el tratamiento oportuno y establecer el pronóstico. Es importante el seguimiento de pacientes con artrogriposis múltiple congénita, con especial atención en la manifestación de contracturas progresivas dolorosas, hiperpigmentación en estas zonas o protrusiones óseas, además de la coexistencia de nódulos y lesiones dérmicas, pues no representan datos característicos de la enfermedad. La difusión de este caso contribuye a la divulgación del síndrome de fibromatosis hialina, con la finalidad de establecer el diagnóstico, tratamiento y pronóstico oportunos y que los pacientes tengan mejor calidad de vida.
REFERENCIAS
1. Maral R, et al. Systemic hyalinosis with heterozygous cmg2 mutations. a case report and review of literature. J Dermatopathol 2016;38:e60-e63.
2. Prabhas PG, et al. Infantile systemic hyalinosis. Indian Pediatr 2012;49:62-63.
3. Rahman N, et al. The gene for juvenile hyaline fibro- matosis maps to chromosome 4q21. Am J Hum Genet 2002;71(4):975-80.
4. Youssefian L, et al. The genetic basis of hyaline fibromatosis syndrome in patients from a consanguineous background: a case series. BMC Med Genet 2018;18(87):1-5.
5. Haidar Z, et al. Diagnosis implications of the whole genome sequencing in a large Lebanese family with hyaline fibromatosis syndrome. BMC Genet 2017;18(3):1-13
6. Bürgi J, et al. CMG2/ANTXR2 regulates extracellular collagen VI which accumulates in hyaline fibromatosis syndrome. Nat Commun 2017;8:15861.
7. Casas D, et al. Hyaline fibromatosis syndrome: clinical up- date and phenotype- genotype correlations. Hum Mutat 2018;39(12):1752-1763.
8. Krishnamurthy J, et al. Juvenile Hyaline Fibromatosis. Indian J Dermatol 2011;Nov;56(6):731-3.
9. Marques SA, et al. Hyaline fibromatosis syndrome: Cuta- neous manifestations. An Bras Dermatol 2016;91(2):226-9.
10. Tzellos TG, et al. Differential expression of matrix metallopro- teinases and proteoglycans in juvenile hyaline fibromatosis. J Dermatol Sci 2011;61:94-100.
11. Bradao FV, et al. Case for diagnosis. An Bras Dermatol 2009;84(6):677-9.
12. Schussier E, et al. Protein-losing enteropathy and joint con- tractures caused by a novel homozygous ANTXR2 mutation. Adv Genomics Genet 2018;8:17-21.
13. Kan AE, et al. Juvenile hyaline fibromatosis: an expanded cli- nicopathologic spectrum. Pediatr Dermatol 1989;6(2):68-75.
14. .Gupta LK, et al. Juvenile hyaline fibromatosis in siblings. Dermatol Venereol Leprol 2005;71(2):115-8.
15. Bordela MT, et al. Casos breves: fibromatosis hialina juvenil. Actas Dermosifiliogr 2004; 95(1):54-7.
16. Shieh JTC, et al. Hyalinosis, inherited systemic. En: Adam MP, et al, editors. Seattle: University of Washington, 1993; 2018.
17. Nischal KC, et al. Juvenile hyaline fibromatosis. J Postgrad Med 2004;50(2):125-6.
18. Hanks S, et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 2003;73 (4):791-800.
19. Kalgaonkar PS, et al. Juvenil hyaline fibromatosis-a rare au- tosomal recessive disease. J Clin Diagn Res 2017;11(7):SD04- SD06.
20. Álvarez P, et al. Abordaje clínico y diagnóstico de la artro- griposis. Acta Pediatr Mex. 2019;40(1):44-50.
21. Rahvar M, et al. Systemic hyalinosis with heterocygous CMG2 mutations. A case report and review of literature. Am J Dermopathol 2016;38:e60-63.
22. Nofal A, et al. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: A unifying term and a proposed grading system. J Am Acad Dermatol 2009;61(4):695-700.
23. Soni JP, et al. infantile systemic hyalinosis: novel founder mutation in the initiation codon among “malis (Farmers)” in Jodhpur. Indian J Pediat. 2016;83(11):1241- 1345.
24. Mantri MD, et al. Hyaline Fibromatosis Syndrome: A rare inherited disorder. Indian J Dermatol 2016; 61(5):580.
25. Lim AA, et al. Juvenile hyaline fibromatosis: report of a case and comparison with infantile systemic hyalinosis. J Oral Maxillofac Surg 2005;63(2):271-4.
26. Castiglione D, et al. Hyaline fibromatosis syndrome (juvenile hyaline fibromatosis): whole-body MR findings in two siblings with different subcutaneous nodules distribution. Skeletal Radiol 2018;47(3):425-431.
27. Tang, et al. Juvenile hyaline fibromatosis: report of a rare case at an advanced stage with osteosclerosis and scoliosis. Int J Dermatol 2016;55(8):903-5.
28. Abduljabbar MH. A case report of juvenile hyaline fibro- matosis. J Dermatol 2014;18:38-42.
Notas de autor
marivi_cervera@yahoo.es