CASOS CLÍNICOS
Enfermedad de Castleman variante multicéntrica: reporte de caso
Castleman's disease multicentric variant: case report

Enfermedad de Castleman variante multicéntrica: reporte de caso
Revista Científica Ciencia Médica, vol. 24, núm. 2, pp. 167-172, 2021
Universidad Mayor de San Simón
Recepción: 18 Abril 2021
Aprobación: 20 Octubre 2021
Resumen:
La enfermedad de Castleman es un reto diagnóstico que se debe tener presente en pacientes con linfadenopatías, debido a que pertenece a un grupo heterogéneo de trastornos linfoproliferativos, fue descrita por Benjamín Castleman en 1956, es una patología poco frecuente, de etiopatogenia aún no establecida, en el que intervienen varios factores como ser: la estimulación antigénica viral,VHH-8,VIH, inmunodeficiencias, elevados niveles de IL-6 que llevan a un proceso inflamatorio lo que desencadena una sintomatología diversa, el diagnóstico definitivo se realiza mediante biopsia excisional. Se presenta el caso clínico de una paciente femenina de 41 años de edad, que acude al servicio de emergencias con cuadro clínico de aproximadamente I año de evolución caracterizado por presentar pérdida de peso progresivo, adenopatías palpables, evidencia de proceso linfoproliferativo y esplenomegalia observados en la tomografía, VIH negativo, anemia y compromiso del estado general.
Palabras clave: Enfermedad de Castleman, Hiperplasia angiofolicular linfoide, trastornos linfoproliferativos, linfadenopatía.
Abstract:
Castleman's disease is a diagnostic challenge that must be taken into account in patients with lymphadenopathy, because it belongs to a heterogeneous group of lymphoproliferative disorders, it was described by Benjamin Castleman in 1956, it is a rare pathology, of still unknown etiopathogenesis, In which several factors intervene such as viral antigenic stimulation, HHV-8, HIV, immunodeficiencies, high levels of IL-6 that lead to an inflammatory process that produces diverse symptoms, the definitive diagnosis is made with excisional biopsy. We present the clinical case of a 41-year-old female patient who came to the emergency department with a clinical picture of approximately I year of evolution characterized by progressive weight loss, palpable lymphadenopathy, evidence of lymphoproliferative process and splenomegaly in the tomography, HIV negative, anemia and compromise of the general state.
Keywords: Castleman's disease, Angiofollicular lymphoid hyperplasia, Lymphoid hyperplasia, Lymphoproliferative disorders, Lymphadenopathy.
INTRODUCCIÓN
La enfermedad de Castleman (EC) es un reto diagnóstico para médicos, radiólogos y patólogos debido a que puede presentarse con amplia o muy limitada sintomatología, además de ser una patología poco frecuente por lo que debemos considerarla cuando estamos ante un paciente con procesos linfoproliferativos1.
La enfermedad de Castleman, también llamada proceso linfoproliferativo o grupo heterogéneo de trastornos linfoproliferativos no neoplásicos, es una entidad poco frecuente, que fue descrita por Benjamín Castleman en 1956, con una gran variedad de localizaciones en las diversas cadenas ganglionares que existen en nuestro organismo2,3.
Es también conocida en la literatura como hiperplasia angiofolicular linfoide, hiperplasia de nódulo linfático gigante, hamartoma de ganglios linfáticos, linfoma benigno, linforreticuloma folicular, hiperplasia linfoide hialinizante, angiomatosis linfoidea3,4.
El presente artículo describe los principales hallazgos clínicos y analíticos sobre un caso de Enfermedad de Castleman, además se incluye una revisión sobre esta patología que resalta los puntos importantes y el reto diagnóstico que esta enfermedad representa.
PRESENTACIÓN DEL CASO
Paciente femenina de 41 años de edad acude al servicio de emergencias del Hospital San Vicente de Paul en febrero de 2021, con cuadro clínico de aproximadamente 1 año de evolución, a la anamnesis la paciente refiere pérdida de peso progresivo de aproximadamente 15 kilos en los últimos seis meses, además presentó astenia, adinamia, hiporexia, caída de cabello, alzas térmicas, deposiciones líquidas, mialgias, náuseas y vómitos durante los últimos tres meses, por lo cual requirió internación en dos ocasiones en un hospital privado, el cuadro se exacerbó en los últimos dos días con malestar general, hiporexia, cefalea, fiebre, dolor abdominal, deposiciones líquidas, náuseas y vómitos en varias oportunidades.
No refiere patologías de base, ni alergias, tampoco consumo de alcohol, tabaco o sustancias controladas, sin antecedentes hereditarios de enfermedades neoplásicas.
Al examen físico: paciente en mal estado general, consciente, orientada en lugar, tiempo y espacio, con los siguientes signos vitales, frecuencia respiratoria de 20rpm, temperatura de 38.2°C, presión arterial de 110/65 mmHg, frecuencia cardiaca de 91 lpm.
En cabeza se observa cuero cabelludo con múltiples aéreas desprovistas de cabello, se evidencia palidez mucocutanea, mucosa yugal y lengua secas; en región cervical se palpa nódulos móviles, de consistencia blanda, de bordes regulares, de aproximadamente un centímetro; en región axilar derecha y región inguinal derecha se palpa dos nódulos con las mismas características anteriormente descritas, tórax sin particular, pulmones sin ruidos sobre agregados, corazón normofonetico, abdomen plano, dolor leve a la dígito presión en región de epigastrio y mesogastrio, ruidos hidroaereos positivos normoactivos; en extremidades se evidencia signo del pliegue positivo.
Por lo que se ingresa a la paciente a salas de internación con el diagnóstico de deshidratación y síndrome febril de probable origen neoplásico.
Interconsultas y otros estudios: la paciente porta exámenes previos del mes de enero que ponen en evidencia anemia, hierro sérico bajo, leucocitosis, plaquetopenia, incremento de la velocidad de eritrosedimentación (VES), transaminasas elevadas (TGP, GOT), fosfatasa alcalina elevada (FAL), proteína C reactiva (PCR) elevada, procalcitonina y ferritina elevados, VIH NEGATIVO, COVID-19 NEGATIVO (Ver tabla N°1).
| ESTUDIO | 08/01/21 | 10/01/21 | 13/01/21 | 15/01/21 | 26/01/21 | 16/02/21 | 19/02/21 | 30/04/21 |
| ERITROCITOS | 3,3 | 4,0 | 4,2 | 3,7 | 3,8 | 2,9 | 4,2 | 2,8 |
| 4,2 - 5,5 millones | ||||||||
| LEUCOCITOS | 23 270 | 16 390 | 7 270 | 6 510 | 8 580 | 12 100 | 13 800 | 29 000 |
| 4 500 - 8 000 | ||||||||
| HEMOGLOBINA | 6,6 g/dl | 12,1 | 11,9 g/dl | 10,9 | 11,4 | 8,3 | 12,6 | 7,6 |
| 12 -16 g/dl | ||||||||
| HEMATOCRITO | 27,1 % | 38 | 37,6% | 35 | 36% | 25 | 38 | 24 |
| 40 - 52% | ||||||||
| PLAQUETAS | 194 000 | 117 000 | 85 000 | 126 000 | 236 000 | 177 000 | 90 000 | 270 000 |
| 140000-420000 mm3 | ||||||||
| SEGMENTADOS | 81% | 73 | 63 | 74% | 93% | 88% | 91% | |
| 42 - 75% | ||||||||
| LINFOCITOS | 11% | 3 | 21 | 12% | 7% | 12 | ||
| 20 - 40 % | ||||||||
| GLICEMIA | 80 mg/dl | 88 | 50 | 64 | 102 | |||
| 80 - 110 mg/dl | ||||||||
| UREA | 35 mg/dl | 14 | 79 | 46 | 69 | |||
| 10 -50 mg/dl | ||||||||
| CREATINIA | 1,1 mg/dl | 1,1 | 2,1 | 1,6 | 2,6 | |||
| 0,7 - 1,4 mg/dl | ||||||||
| GOT 8 - 31 U/L | 45 U/L | 46 | 40 | 88 | ||||
| GPT 3 - 32 U/L | 41 U/L | 29 | 25 | 49 | ||||
| FAL | 705 U/L | 1856 | 185 | 543 | ||||
| 64 - 306 U/L | ||||||||
| COVID - 19 | NEGATIVO | NEGATIVO | ||||||
| LATEX R.A | NEGATIVO | |||||||
| PCR | 96 mg/L | 96 mg/L | POSITIVO | |||||
| (POSITIVO >6 mg/L) | ||||||||
| HIV I-II PRUEBA RA- | NO RE- | |||||||
| PIDA | ACTIVO | |||||||
| GRUPO, FACTOR RH | A Rh + | A RH + | ||||||
| PROCALCITONINA | 2,0 ng/ml | |||||||
| (<0,5 ng/ml) | ||||||||
| SODIO | 135 mEq/l | 136 | 133 | 134 | ||||
| 135 - 145 mEq/l | ||||||||
| POTASIO | 4,4 mEq/l | 2,8 | 2,5 | 2,7 | ||||
| 3,5 - 5,5 mEq/l | ||||||||
| CLORO | 99 mEq/l | 95 | 94 | 95 | ||||
| 98 - 108 mEq/l |
Además porta informe de anatomía patológica de enero de 2021 de biopsia de lesión en piel de rodilla izquierda y de ganglio de región cervical izquierda que reporta: VASCULITIS DE VASOS DE PEQUEÑO CALIBRE (LEUCOCITOCLÁSTICA) en lesión de rodilla izquierda e HIPERPLASIA REACTIVA en ganglio linfático, se realiza estudio inmunohistoquímico que reporta: GANGLIO LINFÁTICO CON HIPERPLASIA FOLICULAR DE TIPO REACTIVA (NO NEOPLÁSICO) Y LINFADENITIS DERMATOPÁTICA.
En febrero de 2021 se le solicita TAC TÓRAX, ABDOMEN Y PELVIS CON CONTRASTE donde se evidencio conglomerados ganglionares mediastinales, retroperitoneales, para aórticos, para cavo aórticos, iliacos inguinales, axilares, con realce al medio de contraste en probable relación a proceso linfoproliferativo y esplenomegalia (Ver figuras 1 y 2).
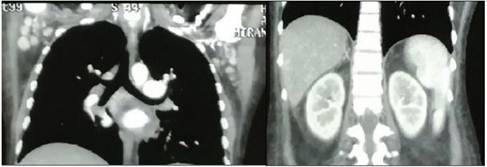
Figura 1
TAC DE TORAX ABDOMEN CON CONTRASTE Presencia de conglomerados ganglionares con realce al medio de contraste a nivel de ganglios axilares bilaterales 20 27 mm y esplenomegalia
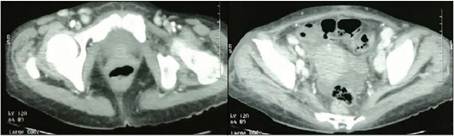
Figura 2
TAC DE PELVIS CON CONTRASTE: Presencia de conglomerados ganglionares con realce al medio de contraste a nivel de ganglios inguinales superficiales y profundos (15 - 20 mm) e iliacos (20-23 mm).
Estudios de laboratorio de febrero de 2021 reportan: anemia, leucocitosis, plaquetopenia, tiempo de protrombina prolongado, aumento de urea y creatinina, PCR positiva, disminución de los electrolitos, COVID-19 NEGATIVO (ver tabla N°1), examen general de orina sin evidencia de infección urinaria, reacción de vidal NEGATIVA.
Se realizó hidratación endovenosa con corrección del desequilibrio hidroelectrolítico, se inició antibioticoterapia y tres días posteriores a la estabilización de la paciente se inició transfusión sanguínea con 4 concentrados de glóbulos rojos (2 por día), posterior a esto se solicitó interconsulta con el servicio de cirugía y se realizó biopsia de ganglio inguinal derecho que reportó hiperplasia linfoide reactiva, patrón hiperplásico con áreas de hialinosis, centro folicular con células dendríticas que corresponden a Enfermedad de Castleman (ganglio linfático con alteración de la arquitectura a expensas de hiperplasia folicular, células dendríticas, linfocitos en la periferia organizados de forma concéntrica, se identifica espacios interfoliculares con incremento de la trama vascular mezclada con linfocitos maduros).
Se solicitó interconsulta con cirugía oncológica para manejo, seguimiento y tratamiento adecuado de la paciente y se dio de alta después de estabilizar y mejorar el estado general de la paciente.
DISCUSIÓN
La enfermedad de Castleman es una entidad rara o poco frecuente, no se tienen muchos datos sobre su epidemiologia e incidencia, se estima que existen 21 casos por millón de habitantes en Estados Unidos; en su clasificación histológica encontramos cuatro variantes entre las cuales tenemos la hialinovascular (80-90%) predominantemente asintomática y localizada; de células plasmáticas o plasmocelular (10%) con sintomatología sistémica en la mayoría de los casos, además de ubicación multicentrica; mixta en la cual se observa características de los dos antes mencionados; la variante plasmablastica en casos agresivos de enfermedad de Castleman multicéntrica asociada a virus herpes humano 8 (VHH-8) y al síndrome de POEMS, esta enfermedad se presenta en la cuarta y quinta década de la vida, con aparente predominancia por el sexo femenino1,2,6.
La variedad Hialinovascular se caracteriza por presentar hiperplasia folicular, zona del manto gruesa e importante y vascularización atípica interfolicular, además se puede observar una capa concéntrica de linfocitos que rodean los centros foliculares que dan la apariencia de capas de cebolla, que representa la imagen característica de la variante hialinovascular, además se puede evidenciar degeneración hialina, los centros germinales son hipocelulares, constituidos predominantemente por células dendríticas, células endoteliales, células reticulares y algunas células B 4,5
En la variedad plasmocelular se puede evidenciar folículos linfoides atróficos o hiperplásicos, expansión de la paracorteza con numerosas células plasmáticas maduras en el espacio interfolicular, folículos linfoides de gran tamaño con centros germinales prominentes6.
Según su localización la EC es: unicentrica (ECU), en una región ganglionar localizada y generalmente de buen pronóstico y multicentrica (ECM), afecta ganglios de regiones diferentes del cuerpo por esto se la denomina diseminada o sistémica con alta morbilidad y mortalidad3.
La localización más frecuente donde se encuentra la EC varía, pero según diversos estudios el mediastino (40-60%) es la principal localización, además se presenta en cuello, abdomen, retroperitoneo, pelvis, axilas y en cualquier ganglio o cadena ganglionar presente en nuestro cuerpo 11,12.
La etiopatogenia no es clara, sin embargo existen diversos factores que pueden ser el punto de partida como la estimulación antigénica crónica de origen viral, alteraciones en la regulación de factores de crecimiento, VHH-8, VIH, inmunodeficiencias, inflamación crónica, infección por el virus de Ebstein Barr 3,4.
La interleucina 6 (IL-6) en niveles elevados induce la proliferación de células B, diferenciación de los linfocitos B y T, secreción de hepcidina, lo que estimula la síntesis hepática de reactantes de fase aguda que a su vez producen un espectro inflamatorio que causa la sintomatología, además induce la expresión del factor de crecimiento del endotelio vascular (VEGF) que podría hipervascularizar regiones germinales de los ganglios linfáticos, mecanismo que aún no está bien definido 10,11.
El VIH debido a su gran capacidad replicativa, la estimulación constante de los linfocitos B y la inmunodepresión son factores etiopatogenicos, además el VHH-8 estimula la producción de IL-6 viral similar a la IL-6 humana, lo que produce mayor secreción de IL-6 humana perpetuando el ciclo inflamatorio7,8
Puede ser asintomática en la ECU, pero generalmente la ECM va acompañada de sintomatología sistémica diversa y con diferente intensidad como astenia, adinamia, pérdida de peso, mal estar general, fiebre, sudoración nocturna, anorexia, debilidad, linfadenopatías dolorosas, además puede encontrarse hepatomegalia, esplenomegalia, ascitis, edema, derrames pleural y pericárdico7,8. También se evidenció trombocitopenia, anemia, hipoalbuminemia, hipergammaglobulinemia, pancitopenia y falla orgánica 8,9.
El diagnóstico definitivo se realiza con biopsia excisional y análisis histopatológico, la punción y aspiración con aguja fina no se recomienda y los estudios de imagen como la tomografía nos muestran regiones con áreas procesos linfoproliferativos además de hepato- esplenomegalia 9,10. El diagnóstico diferencial se ejecuta con timomas, enfermedad reumatoide, teratomas, carcinomas, linfoma, tuberculosis, sarcoidosis, enfermedades autoinmunitarias 11.
El tratamiento de la forma localizada es la exéresis completa quirúrgica del ganglio linfático afectado, en ambas variantes histológicas, con remisión completa en la mayoría de los casos y aparentemente excepcionales recurrencias, en la forma multicéntrica, aún no hay un consenso sobre un tratamiento definido, sin embargo se plantean tratamientos diversos, entre ellos la resección quirúrgica, combinado con quimioterapia y radioterapia, además del uso de esteroides 12.
REFERENCIAS
1. Bellolio J Enrique, Tapia Óscar, Iriarte C Mª José. Enfermedad de Castleman, un diagnóstico diferencial que no se debe olvidar en los tumores de mediastino: Caso clínico. Rev. méd. Chile [Internet]. 2014 [citado 2021 Abr 03]; 142( 6 ): 782-785. Disponible en: https://scielo.conicyt.cl/scielo.php?script=sci_arttext&pid=S0034-98872014000600013&lng=es. http://dx.doi.org/10.4067/S0034-98872014000600013.
2. K. de Aguiar Quevedo, C. Jordá Aragón, N. Mancheño Franch, G. Sales Badía, V. Calvo Medina, et al. Enfermedad de Castleman: dos variantes, dos pacientes. Revista de Patología Respiratoria [Internet]. 2011 [citado 2021 Abr 03]; 14( 4 ): 138-142. Disponible en: https://www.elsevier.es/es-revistarevista-patologia-respiratoria-318-articulo-enfermedadcastleman-dos-variantes-dos-X1576989511589680https://doi.org/10.1016/S1576-9895(11)70126-6.
3. Sevilla-Lizcano DB, Frias-Soria CL, Ortiz-Hidalgo C. Enfermedad de Castleman. Análisis histopatológico e inmunohistoquímico de treinta y nueve casos. Gac Med Mex. [Internet] 2017 [citado 2021 Abr 03]; 153(5):550-558. Disponible en: https://www.anmm.org.mx/GMM/2017/n5/GMM_153_2017_5_550-558.pdf
4. Hollman PL, Bianchi F, Chaves L.A, PerézFirpo M, Ghiglione P. Enfermedad de castleman: un caso de presentación atípica: Rev. Arg. Res. Cir. [Internet] 2017 [citado 2021 Abr 03]; 22 (2) 19-20. Disponible en: https://revistaaamrcg.org/wp-content/uploads/2020/08/Rev.-Arg.-Res.-Cir.-2017_2.pdf#page=19
5. Zapata-Bonilla SA, López-Vargas R, Scherling-Ocampo AA, et al. Enfermedad de Castleman Multicéntrica tipo hialino-vascular en paciente inmunocompetente. Gac Med Mex. [Internet] 2015 [citado 2021 Abr 06]; 151(5):648-654. Disponible en: https://www.anmm.org.mx/GMM/2015/n5/GMM_151_2015_5_648-654.pdf
6. González García A, Moreno Cobo MA, Patier de la Peña JL. Diagnóstico y tratamiento actual de la enfermedad de Castleman. Revista Clínica Española. [Internet] 2015 [citado 2021 Abr 06]; 216 (3): 146-156. Disponible en: https://www.sciencedirect.com/science/article/pii/S0014256515002829https://doi.org/10.1016/j.rce.2015.11.008
7. Rojo Abecia M, Pérez Morera A, Roldán Cortés D, Ruiz De la Hermosa A, Yagüe Rigopolou N, Román García de León L, Paseiro Crespo G. Enfermedad de Castleman unicéntrica: a propósito de un caso. Cir Andal. [Internet] 2021 [citado 2021 Abr 03]; 32(1):70-73. Disponible en: https://www.asacirujanos.com/admin/upfiles/revista/2021/Cir_Andal_vol32_n1_14.pdf
8. Dimas E Hernández, Mario Comegna, José López, Marie L García. Enfermedad de Castleman multicéntrica asociada a infección por virus de inmunodefificiencia humana, reporte del primer caso venezolano. Revista Venezolana de Oncología. [Internet] 2016 [citado 2021 Abr 03]; 28 (1): 52-56. Disponible en: https://www.redalyc.org/journal/3756/375643222007/html/index.html
9. Belli MHR, Cauich EE, Cih OGA, et al. Manifestaciones neurológicas de la enfermedad de Castleman: Reporte de caso. Rev Mex Neuroci. [Internet] 2016 [citado 2021 Abr 06]; 17(4):116-124. Disponible en: https://www.medigraphic.com/pdfs/revmexneu/rmn-2016/rmn164m.pdf
10. Guzmán-Fernández MR, Campoy-García F, Pereiro-Sánchez M, Sastre-Moral JL. Espectro clínico de la Enfermedad de Castleman. Galicia Clin [Internet] 2014 [citado 2021 Abr 06]; 75 (4): 191-194. Disponible en: https://galiciaclinica.info/PDF/30/569.pdf
11. Muñoz-Lumbreras RG, Morales-Ordaz O, GilGarcía F, Lujano-Pedraza H, Valdéz-Colín J, MichelRamírez JM y col. Enfermedad de Castleman con manifestación inusual en el seno y parénquima renal. Reporte de un caso. Rev Mex Urol. [Internet] 2018 [citado 2021 Abr 06]; 78(1):62-66. DOI: https://doi.org/10.24245/revmexurol.v78i1.1457
12. Vieira Gusmão L, Dandara Correia de Pinho P, Moura Guimarães Dota V.O, da Silva Boa Sorte E.M, Malheiro Ferreira L, Chaves de Aguiar Filho J.A, Fernandes Barbosa G.E, Janes dos Reis C. Doença de Castleman axilar: Relato de Caso. Revista Eletrônica Acervo Saúde. [Internet] 2020 [citado 2021 Abr 06]; 12(1): 1-8. Disponible en: https://doi.org/10.25248/reas.e1801.2020
Notas
_______
Notas de autor
Enlace alternativo
http://www.scielo.org.bo/scielo.php?script=sci_arttext&pid=S1817-74332021000200167&lng=es&nrm=iso (html)