Resumo: O estudo e desenvolvimento de polímeros para aplicação nas áreas biomédica e farmacêutica têm aumentado devido às suas propriedades peculiares que contribuem para a melhoria da qualidade de vida, como por exemplo, os polímeros usados em medicina regenerativa e sistemas de liberação de fármacos. O desenvolvimento de novos materiais baseados em polímeros depende desde os métodos de síntese, extração ou composição desses materiais até os estudos da influência de suas estruturas e propriedades em aplicações específicas, entre outras. Esta revisão descreve o uso de alguns de polímeros novos e convencionais com potencial para aplicação nas áreas farmacêutica e biomédica, enfatizando as principais propriedades que os tornam aplicáveis.
Palavras-chave:BiopolímeroBiopolímero,medicina regenerativamedicina regenerativa,liberação de fármacosliberação de fármacos.
Abstract: The study and development of polymers for pharmaceutical and biomedical use has been increasing due to their peculiar properties that contribute for the improvement of the life quality, such as the polymers used in regenerative medicine and in drug release systems. The development of new polymer based materials and its composites depends on several steps, such as the synthesis approach, the extraction, the composition, the influence of their properties on the specific applications, and others. This review describes the use of conventional and new polymers with potential application in pharmaceutical and biomedical fields, highlighting the properties that allow them to be useful for such purposes.
Keywords: Biopolymer, regenerative medicine, drug delivery.
Biopolímeros: aplicações farmacêutica e biomédica
Universidade Estadual Paulista Júlio de Mesquita Filho

Este trabalho está sob uma Licença Internacional Creative Commons Atribuição 4.0.
A demanda de novos materiais para a terapia celular, medicina regenerativa e liberação de fármacos é crescente em função do decréscimo do tempo de recuperação e a melhoria da qualidade de vida de pacientes beneficiados por tais sistemas. Sistemas para carreamento e liberação de fármacos e materiais para substituição permanente ou temporária de tecidos injuriados são exemplos de aplicações crescentes na área biomédica. Os polímeros degradáveis in vivo, naturais ou sintéticos, são outro exemplo a ser citado para aplicação em substituição temporária de tecidos. O desenvolvimento de novos materiais para uso nestas áreas sofreu grandes evoluções nas últimas décadas, porém, apesar de encontrar diversos sistemas e materiais com aplicação clínica, existe muito a ser explorado em busca daqueles com propriedades mais próximas das ideais1,2. As propriedades dos polímeros, por sua vez, dependem da sua estrutura química.
A degradação desses materiais no interior de organismos animais leva conforto ao paciente no sentido de evitar uma cirurgia para remoção do material implantado. Nesse contexto, a estrutura química dos materiais está diretamente relacionada com sua taxa de degradação e ainda necessita ser estudado a respeito da relação estrutura química/degradação. Os produtos de degradação desses materiais também devem ser considerados durante a avaliação de um biomaterial, pois não devem ser tóxicos ou causar quaisquer tipos de efeitos não desejados ao paciente. Materiais não completamente degradáveis, porém, biocompatíveis, são também de grande valia para as áreas em questão, visto que podem compor sistemas inovadores, conforme apresentado no decorrer deste texto2,3.
Dentro desse contexto, este trabalho descreve as principais propriedades e aplicações de alguns polímeros que vêm ganhando destaque nas áreas farmacêutica e biomédica, a iniciar-se pela fibrina, um polímero natural de função e aplicação já bem estabelecidas em procedimentos médicos. Em seguida, serão abordados a celulose bacteriana, pectina, amido resistente, quitosana, goma gelana, ácido poliláctico e policaprolactona, polímeros que se encontram em estágio de pleno desenvolvimento para futuras aplicações práticas nas áreas aqui abordadas.
Fibrinogênio e fibrina constituem proteínas que apresentam funções importantes em vários processos biológicos, como coagulação sanguínea, fibrinólise, interações celulares, resposta inflamatória e processo de cicatrização4.
O fibrinogênio consiste em uma glicoproteína solúvel de 340 kDa, normalmente presente no plasma sanguíneo em concentração aproximada de 2,5 g L-1 que, por ação proteolítica da trombina, é convertido em fibrina4,5, sendo uma proteína bastante heterogênea devido a variações na proteólise parcial, fosforilação ou sulfatação de resíduos de aminoácidos, polimorfismo genético e splicing alternativo6.
Estruturalmente, o fibrinogênio é formado por cadeias αα’, ββ’ e 2 cadeias g unidas por 29 ligações bissulfeto. As regiões C-terminais das cadeias ββ’ e g formam os domínios D, e as regiões N-terminais das cadeias restantes, formam o domínio E. Os domínios D se encontram conectados ao domínio E por dois segmentos de α- hélices contorcidas (Figura 1). As regiões Cterminais αα’ são globulares e encontram-se ligadas ao domínio E7,8.
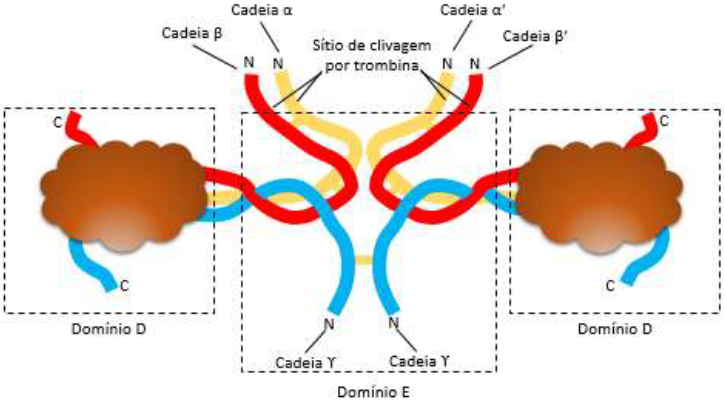
Figura 1
Estrutura do Fibrinogênio.
O processo de formação da fibrina tem início com a clivagem proteolítica da região N-terminal de αα’e ββ’, pela trombina, com a liberação dos fibrinopeptídeo A (FPA) e B (FPB), sendo que a clivagem do FPA induz a polimerização em protofibrilas5, enquanto FBP é clivado mais lentamente do que FPA pela trombina9. A ligação cruzada entre os monômeros de fibrina é estabilizada pelo fator XIIIa (Figura 2) que proporciona melhoria nas propriedades mecânicas e bioquímicas da fibrina e promove resistência à fibrinólise10.
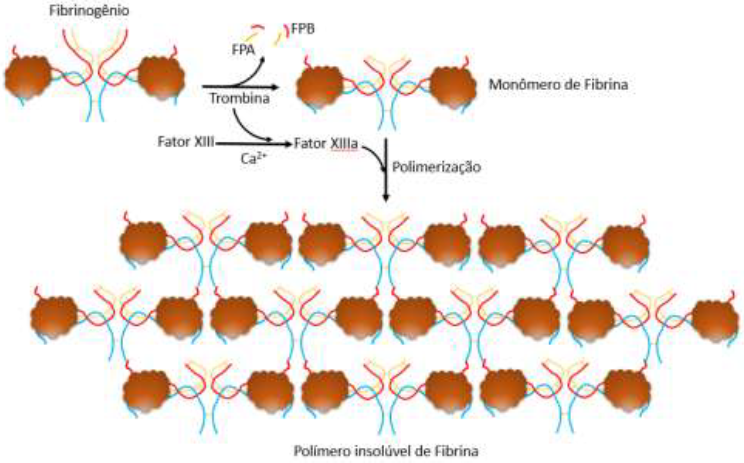
Figura 2
Polimerização e formação do polímero insolúvel de fibrina.
Outra propriedade importante da fibrina é a biodegradabilidade determinada pela ação da plasmina, originária do plasminogênio, por ação do Fator Ativador de Plasminogênio Tecidual (AP-t) e a uroquinase (Figura 3).
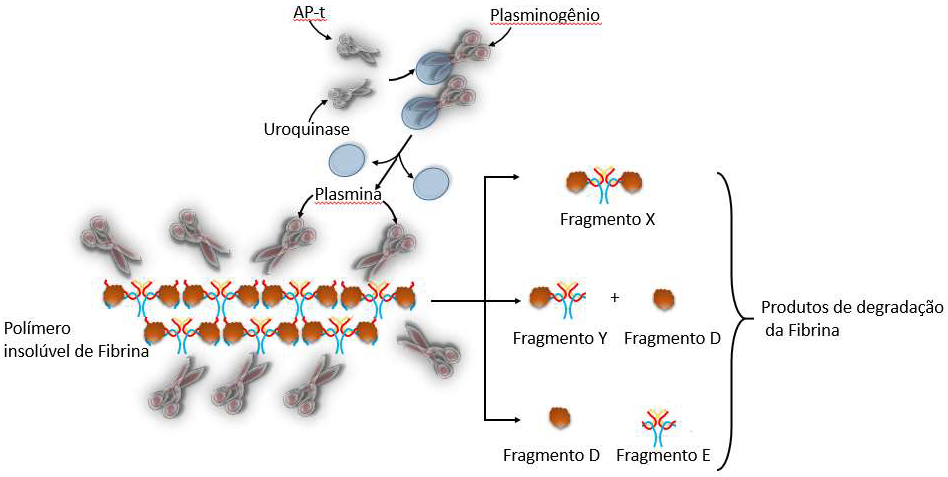
Figura 3
Fibrinólise.
As propriedades bioquímicas e mecânicas apresentadas pela fibrina permitem sua aplicação em medicina e bioengenharia como selante11,12 aprovado pelo Food and Drug Administration (FDA).
O selante de fibrina é um material constituído basicamente por dois componentes, fibrinogênio e trombina, que na presença de cálcio e do fator XIIIa, a trombina é capaz de converter fibrinogênio em fibrina insolúvel, que constitui a forma estável final do agente12. Os relatos da utilização de selantes de fibrina datam de longo tempo, como pinça13, como adesivo14. São várias as aplicações dos selantes de fibrina em especialidades cirúrgicas, como cirurgia geral, cirurgia vascular e cardiovascular, neurocirurgia, cirurgia urológica15, cirurgia plástica e reconstrutiva, incluindo cirurgia de reconstituição de queimaduras, cirurgia craniofacial e cirurgia microvascular16, como cola hemostática17, artroplastia de joelho18,19e cirurgias odontológicas20. Sua vasta aplicação clínica deve-se basicamente às suas propriedades químicas, com destaque para sua capacidade de adesão, biodegradabilidade e biocompatibilidade. Como se pode concluir, selantes de fibrina apresentam múltiplos usos e a realização de novos estudos randomizados in vivo devem resultar em melhorias na assistência intra e pós-operatória ao paciente.
A celulose é um dos biopolímeros mais abundantes da natureza e, embora os vegetais constituam a fonte mais importante desse polímero, ele também pode ser produzido por vários tipos de organismos, incluindo bactérias21,22. Em vegetais, a celulose encontra-se associada a outros componentes como lignina e hemicelulose. A celulose bacteriana (CB) constitui um homopolímero natural (poli -D-glicose) linear com estrutura constituída por unidades de b- D-glicopiranose unidas por ligações glicosídicas do tipo isento de lignina e hemicelulose (Figura 4)23.

Figura 4
Desenho esquemático da diferença entre celulose vegetal constituída de celulose, lignina e hemicelulose e a celulose bacteriana constituída pela celulose pura24.
A CB possui a mesma estrutura química da celulose proveniente de plantas, mas suas fibras de dimensões nanométricas conferem-lhe propriedades distintas. Entre essas propriedades, destacam-se a alta resistência mecânica à tração e a possibilidade de inserção de materiais para obtenção de compósitos no espaço entre as fibras. Recentemente, essas inserções têm sido feitas in situ, durante o cultivo de bactérias do gênero Gluconacetobacter23.
A formação de fibras de celulose é devida à ocorrência de ligações de hidrogênio, responsáveis pela consistente associação entre as macromoléculas lineares de celulose. Estruturalmente, a CB é composta por uma rede tridimensional de nanofibras, mantidas por ligações de hidrogênio com interações intra e interfibrilares (Figura 5) resultando em um hidrogel com elevada resistência mecânica. O primeiro tipo de interação é responsável pela rigidez da cadeia e o segundo pela formação da fibra vegetal25,26.
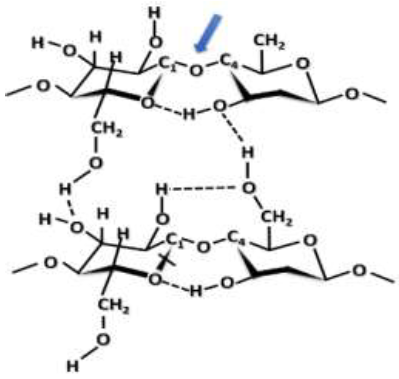
Figura 5
.Estrutura da celulose. As linhas pontilhadas esquematizam as ligações de hidrogênio possíveis e a seta azul, a ligação.
As nanofibras constituem uma estrutura tridimensional com grande quantidade de espaços vazios entre as fibras, criando assim uma área de superfície estendida com geometria variada dos poros (Figura 6) proporcionando propriedades notáveis à CB, incluindo alta resistência mecânica, elevada cristalinidade, alta capacidade de retenção de água, biodegradabilidade, biocompatibilidade e capacidade de ter suas estruturas tridimensionais moldadas durante a síntese23,27.
As membranas de CB também apresentam elevada capacidade de absorção de diferentes espécies iônicas, moleculares ou até mesmo a estabilização de partículas, pois apresentam uma estrutura altamente hidratada. Essa estrutura tem favorecido a utilização da CB como agente de reforço, como molde, na preparação de copolímeros e na formação de redes interpenetradas com elevado grau de intercruzamento28,29.
A CB pode ser produzida por bactérias como Sarcina, Agrobacterium, Rhizobium e Acetobacter30, entre outros micro-organismos. Um destaque especial é dado às bactérias do gênero Gluconacetobacter, especialmente pelas espécies G. xylinus e G. hansenii (Figura 6) devido à sua alta produtividade, utilizando uma variedade de meios de cultivo naturais ou sintéticos, com diferentes fontes de carbono. Desde a sua descoberta, a CB demonstrou ser um biopolímero de grande interesse para aplicação em várias áreas industriais e médicas31,32.

Figura 6
Microscopia Eletrônica de Varredura da G. hansenii e a ultraestrutura da CB secretada pelo micro- organismo.
Bactérias do gênero Gluconacetobacter utilizam uma variedade de substratos carbônicos para sintetizar a celulose, entretanto, a síntese depende do ciclo de pentoses e do ciclo de Krebs23,33. A conversão da glicose, transportada a partir do ambiente externo para o citoplasma, é catalisada por quatro enzimas bacterianas: glucoquinase (responsável pela obtenção da glicose-6-fosfato), fosfoglicomutase (responsável pela catálise da glicose-6-fosfato a glicose-1-fosfato), UDPG- pirofosforilase (responsável pela síntese da UDP- glicose) e a celulose sintase (responsável pela polimerização da celulose a partir da UDP-glicose) (Figura 7). A reação de síntese da celulose bacteriana consome cerca de 10% do ATP gerado no metabolismo bacteriano, dessa forma, utilizando para sintetizar a celulose o metabolismo aeróbico34.
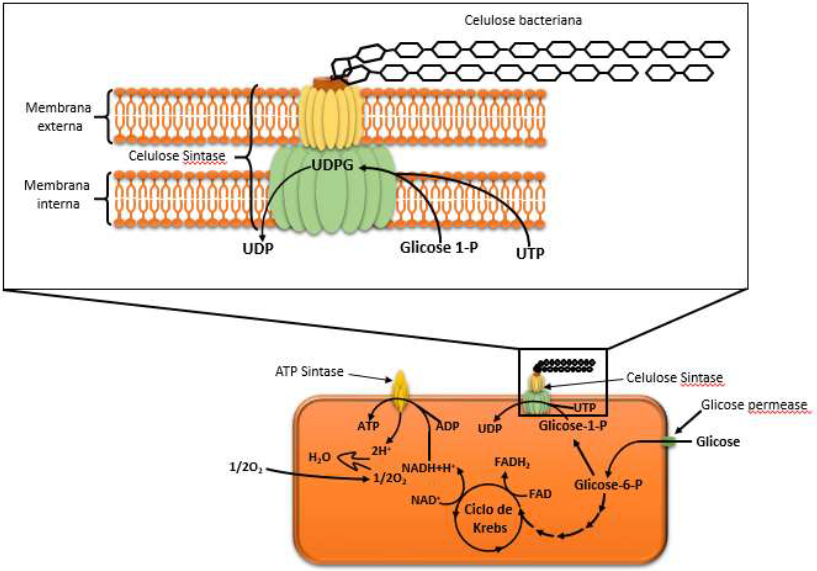
Figura 7
Representação esquemática da síntese da CB.
A degradação da celulose, considerando as fontes vegetais ou bacterianas, acontece através de reações enzimáticas35. A celulose, por ser um polímero complexo e insolúvel, formada por moléculas de glicose, precisa necessariamente, para sua degradação, a ação das celulases36. Na natureza, a hidrólise da celulose ocorre por ação de micro- organismos produtores de celulases, enzimas que hidrolisam a ligação glicosídica.
Segundo Backdahl e colaboradores37, a degradação da CB não está bem esclarecida nem in vitro, nem in vivo. Em animais, a ausência das celulases faz com que sua degradação seja limitada para aplicação na área biomédica. Embora o interesse na área biomédica seja de um material completamente degradável, a otimização e a sincronia do tempo de degradação relacionado às propriedades mecânicas de biomaterial, como a CB, também pode ser vantajoso em relação aos degradáveis38.
A elevada pureza e as propriedades físico- químicas da CB oferecem uma variedade de aplicações potenciais, como por exemplo, em indústrias de alimentos, indústria têxtil e, especialmente na área biomédica como curativos para ferimentos25,32,39, substituto temporário de pele no tratamento de úlceras, queimaduras, lesões e enxertos38, vasos sanguíneos40 e, em particular, como scaffolds para engenharia de tecidos41,42 e matriz para liberação controlada de fármacos43. Diante do exposto, pode-se concluir que membranas de CB apresentam diversas aplicações industriais e médicas e a realização de novos estudos para otimizar a produção com baixo custo tem atraído pesquisadores no mundo todo.
A pectina é um heteropolissacarídeo encontrado principalmente na lamela média e na parede celular primária das plantas, sendo responsável pela manutenção de estruturas e pela sustentação da planta44,45. Ela é encontrada abundantemente na natureza, porém, poucas plantas são utilizadas como fonte desse polissacarídeo, devido ao custo, ao rendimento e ao tempo do processo de extração. As substâncias pécticas normalmente são extraídas a partir de tecidos frescos ou secos de frutas ou vegetais através de diferentes tipos de processos extrativos, tais como extração com água quente, com compostos quelantes e com enzimas. As fontes naturais de pectina que apresentam maior rendimento na sua extração são frutas cítricas, maracujá, toranja e manga; no entanto, esse polissacarídeo tem sido obtido basicamente das cascas de frutas cítricas ou do bagaço da maçã e de resíduos das indústrias que processam sucos44,46,47.
Quimicamente, as pectinas são macromoléculas compostas por homogalacturonana (HG) e ramnogalacturonanas (RG), que são divididas em dois tipos: ramnogalacturonanas І (RG І) e ramnogalacturonanas ІІ (RG ІІ). Ainda existe um quarto domínio denominado xilogalacturonana (XG)45,48.
A HG, descrita como a região lisa ou smooth region, representa cerca de 65% da estrutura total da pectina e é uma cadeia linear constituída de resíduos de ácidos galacturônicos (GalA) unidos por ligações glicosídicas do tipo α-(1→4), parcialmente esterificados com grupamentos metila e acetila46,48,49.
As RG І e RG ІІ são definidas como hairy region ou regiões altamente ramificadas. A RG І compreende 20 a 35% da estrutura da pectina, constituída de unidades repetidas de dissacarídeos de ácido D-galacturônico ligadas α-(1→4) e ramnose α-(1→2), com uma variedade de cadeias laterais de resíduos de arabinose e galactose, ligadas na forma de mono ou oligossacarídeos49.50,51. A RG ІІ, representante da região mais complexa da estrutura da pectina, é composta por uma cadeia principal, com aproximadamente nove ligações de ácido galacturônico com quatro cadeias laterais complexas, compreendendo 12 monossacarídeos distintos ligados por cerca de 20 diferentes ligações glicosídicas52,53. Dentre os açúcares das cadeias laterais incluem-se os açúcares raros, como o ácido acérico, a apiose, o Dha (ácido 3-deoxi-D- lixoheptulosárico) e o K do (ácido 2-ceto-3-deoxi- D-manooctulosônico)53,54,55.
As XG são constituídas por cadeias de HG, parcialmente substituídas na posição O-3 β-D- xilose, e estas podem ser substituídas por uma molécula adicional de xilose gerando um dissacarídeo45,56.
As moléculas de pectina compreendem segmentos lineares de ácido galacturônico, nos quais os grupos carboxílicos podem estar esterificados com metanol (metoxilação). O grau de esterificação (DE) da pectina é definido a partir da proporção dos resíduos de ácido galacturônicometoxilados presentes na molécula. Alguns dos grupos carboxílicos podem se converter em grupos carboxamida, quando a amônia é utilizada no processo de desesterificação, formando pectina amidada. Dessa forma, o DE e o grau de amidação (DA) determinam o conteúdo de ácido carboxílico presente nas cadeias de pectina47,57.
De acordo com o DE, as pectinas são divididas em duas classes: pectinas de alto grau de metoxilação, quando DE > 50%, e pectinas de baixo grau de metoxilação, com um DE < 50%44,58.
O gel de pectina é formado pela reação de reticulação das cadeias de HG, resultando em uma rede tridimensional em que ocorre o aprisionamento de água e outras moléculas46. Os fatores que influenciam a formação do gel e que determinam a ocorrência da geleificação são temperatura, pH, concentração de pectina, concentração de solutos (açúcares) e concentração de íons, tais como o Ca+2 46,47,57. A forma como estes fatores influenciam a geleificação são diretamente proporcionais às propriedades moleculares das pectinas, como massa molar, o grau de esterificação e de amidação, a presença de ésteres de acetila na cadeia de galacturonana e a distribuição de ramnose ao longo da cadeia de pectina46, 47.
Os mecanismos de geleificação são diferentes para as pectinas de alto grau de metoxilação e pectinas de baixo grau de metoxilação47. Nas pectinas de alto grau de metoxilação ocorre a formação de zonas de junção por interações hidrofóbicas e ligação de hidrogênio entre os grupos metoxilados, em pH ácido (< 3,5) e na presença de elevada concentração de açúcares. Nas pectinas de baixo grau de metoxilação, a formação de géis ocorre na presença de cátions divalentes, como íons Ca+2, que agem como uma ponte entre pares de grupos carboxílicos de diferentes cadeias de pectinas, formando zonas de junção em uma ampla faixa de pH (Figura 8)46,47. Esta condição de formação de zonas de junção é relatada através do modelo caixa de ovos (egg box), no qual as cadeias de pectinato são hélices duplas com arranjo antiparalelo, e essas cadeias, por sua vez, formam espaços ou fendas de ligações nas zonas de junção, aprisionando íons cálcio que ligam as cadeias entre si. O mecanismo de geleificação supostamente compreende duas etapas, sendo a primeira a formação de dímeros (egg box) e a segunda a condensação destes dímeros em agregados (egg box sobrepostas)47.
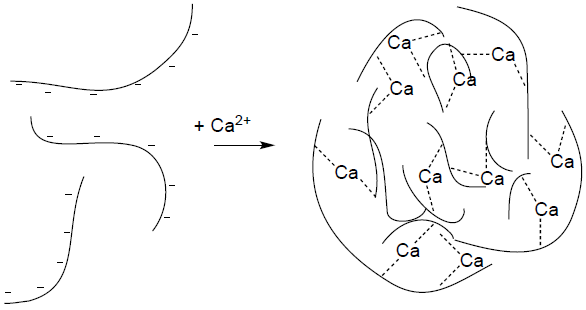
Figura 8
Mecanismo de reticulação da pectina de baixo grau de metoxilação na presença de íons Ca+2 73.
Nos últimos anos, a pectina tem sido amplamente empregada pela indústria de alimentos como agente espessante, geleificante e estabilizante46,59. Dentro da área farmacêutica vem sendo explorada para o desenvolvimento de novos sistemas de liberação de fármacos, devido às propriedades promissoras desse material como baixa toxicidade, biocompatibilidade, degradabilidade e baixo custo60,61. Outra vertente muito explorada é a utilização da pectina como excipiente na obtenção de sistemas de liberação cólon-específica de fármacos, uma vez que em meio ácido permanece como agregados de macromoléculas, além de ser resistente às proteases e às amilases presentes nas porções superiores do trato gastrintestinal (TGI), sendo degradada por enzimas produzidas pela microbiota colônica44,50,57. Além disso, a pectina apresenta reconhecida propriedade mucoadesiva, que agrega significativa vantagem no delineamento de sistemas de liberação controlada de fármacos, devido a capacidade desse material de interagir com a mucina, promovendo um contato mais íntimo entre o sistema e a membrana biológica62,63. Apesar dessas propriedades favoráveis da pectina, o grande desafio da utilização desse polissacarídeo no desenvolvimento de sistemas de liberação controlada de fármacos, é sua elevada solubilidade em meio aquoso, que pode resultar na liberação prematura e indesejável do fármaco nas porções superiores do TGI64,65.
A pectina isolada ou combinada com outros polímeros, naturais ou sintéticos, também denominadas blendas poliméricas, representa uma estratégia racional para a obtenção de materiais com propriedades moduladas, permitindo o desenvolvimento de sistemas inovadores que atendem a necessidades terapêuticas específicas57,66. O desenvolvimento de sistemas orais baseados em misturas de pectina com outros polissacarídeos, bem como o método de reticulação, têm sido amplamente explorados, visando à liberação sítio específica de fármacos no cólon64,66.
Recentemente, a pectina também tem sido investigada na vetorização de genes50, na cicatrização de feridas54,67, no direcionamento (targeting) de fármacos no tratamento do câncer68 e na modificação da superfície de dispositivos médicos69. Além disso, surgiu um grande interesse na utilização da pectina na engenharia de tecidos, principalmente devido à semelhança química desse material com a matriz extracelular de tecidos dos mamíferos54,70. Desta forma, géis de pectina têm sido empregados no desenvolvimento de scaffolds para aplicações em tecido ósseo a fim de estimular a regeneração e promover sua reconstrução44,70. Além disso, a pectina apresenta atividade anti- inflamatória71 e anticarcinogênica72,73. Nesse sentido, a pectina apresenta diferentes propriedades que evidenciam sua importância para o desenvolvimento de novos sistemas de liberação de fármacos, permitindo a busca de aplicações inovadoras de acordo com as diferentes necessidades terapêuticas.
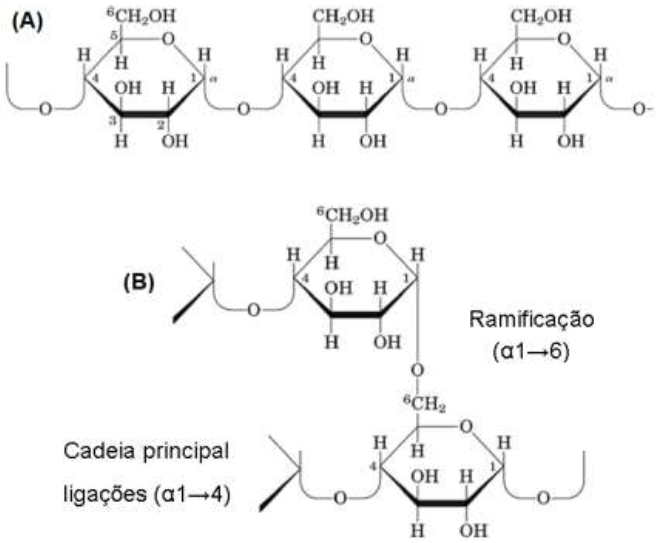
O amido, um dos biopolímeros naturais mais importantes, está presente como principal fonte de energia nos grãos de cereais e de túberculos, incluindo o arroz, o trigo, o milho, a cevada, dentre outros. O polímero se apresenta na forma de grânulos de 1 a 100 μm de diâmetro e é composto principalmente por cadeias de amilose, correspondente à fração linear com ligações glicosídicas α-1,4, e amilopectina, a qual representa um glicano ramificado com ligações α-1,674 (Figura 9). No entanto, de acordo com a espécie vegetal do amido, verifica-se uma grande variação morfológica e estrutural, bem como nas quantidades de amilose, amilopectina, proteína e lipídeo, resultando em diferentes propriedades físico-químicas75,76.
A estruturação do amido no grânulo pode ocorrer de diversas maneiras, as quais são classificadas em 6 níveis77, iniciando-se com ramificações individuais e distribuídas no sentido do comprimento das cadeias (nível 1), as quais podem se organizar e levar à formação de aglomerados (nível 2). Em seguida, formam-se as lamelas cristalinas (ramificações de amilopectina) e amorfas (amilose), em resposta ao empacotamento das cadeias de glicano para formação de duplas hélices (nível 3). A organização das lamelas em camadas de crescimento concêntricas com as lamelas cristalinas e amorfas separadas radialmente corresponde ao grânulo de amido (nível 4), o qual, ao se associar à proteínas e lipídeos, dá origem ao endosperma (nível 5), seguido pelo nível final (nível 6), o qual corresponde ao grão de amido inteiro de 1 mm de tamanho, incluindo estruturas bastante organizadas.
O amido tem sido alvo de extensa pesquisa nas últimas décadas nas áreas alimentícia, cosmética e de medicamentos, merecendo destaque a última, como excipiente de formas farmacêuticas sólidas, devido às suas importantes vantagens como a biodegradabilidade exercida pela microbiota colônica, atoxicidade, baixa reatividade com substâncias ativas medicamentosas e baixo custo78,79. Contudo, a aplicação do amido no desenvolvimento de novos sistemas de liberação de fármacos requer propriedades físicas e químicas específicas, que muitas vezes o amido nativo não apresenta, como, por exemplo, baixa solubilidade em água, baixo intumescimento e alta resistência contra a degradação enzimática80. Nesse sentido, a modificação do amido através de métodos físicos, químicos e/ou enzimáticos torna-se essencial para a otimização de suas propriedades78.
Um tipo de amido modificado que vem sendo bastante difundido é o amido resistente (AR), o qual corresponde à fração do amido que não participa da digestão no intestino delgado, sendo fermentado pelas enzimas bacterianas colônicas em ácidos graxos de cadeias curtas, propriedade fundamental para sua aplicação em sistemas de liberação cólon- específica de fármacos81.
De acordo com a natureza do amido, o AR pode ser dividido em 4 subtipos, sendo o AR tipo 1 fisicamente inacessível à digestão devido seu aprisionamento na matriz; o AR tipo 2 que não é gelatinizado, o AR tipo 3 que é o amido retrogradado e o AR tipo 4 que é o amido quimicamente modificado82.
Devido à sua estabilidade térmica e baixa solubilidade, o amido retrogradado (AR tipo 3) tem despertado grande interesse83,84,85. Sua obtenção se dá através do processo de retrogradação, iniciando-se com a etapa de gelatinização, na qual a estrutura semicristalina do amido torna-se amorfa pelo aquecimento em excesso de água, seguido por ciclos térmicos de armazenamento e resfriamento para a recristalização lenta dos componentes do amido em estruturas tridimensionais mais organizadas76, que ocorre pela formação de ligações de hidrogênio e forças de Van Der Waals inter e intramoleculares, no intuito de se alcançar uma forma metaestável de mais baixa energia livre86, (Figura 10).
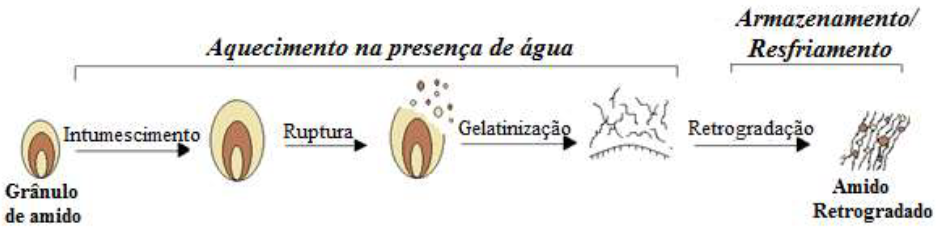
Figura 10
Processo esquemático da obtenção do amido retrogradado.
A alta amilose (amido modificado composto por 70% de amilose) tem sido considerada o material preferido para obtenção de AR65,87, uma vez que é responsável pela formação de uma matriz amorfa onde os cristalitos permanecem embebidos e, então, protegidos da degradação enzimática.
Dispersões filmógenas com elevado teor de AR (~96%) foram obtidas por Meneguin e colaboradores65 a partir da retrogradação de alta amilose na presença de pectina por meio de ciclos térmicos alternados a 4 ºC e 30 ºC durante 16 dias, sendo 2 dias em cada temperatura. Filmes de revestimento foram obtidos pela evaporação do solvente dessas dispersões retrogradadas, dando origem a filmes livres com propriedades mecânicas (resistência à perfuração e alongamento durante a perfuração) e de barreira (permeabilidade ao vapor d'água) adequadas. Além disso, esses filmes demonstraram ser excelentes candidatos para a liberação cólon-específica de fármacos, uma vez que os mesmos se mostraram altamente resistentes contra a degradação quando incubados com α- amilase pancreática, além de terem exibido baixa dissolução (%) em meio ácido.
Recife88 em estudos com comprimidos matriciais de amido retrogradado avaliou a influência do método de retrogradação nas propriedades físico-químicas e de liberação do diclofenaco de sódio, concluindo que ciclos mais curtos (8 dias ao invés de 16) e isotérmicos (4 ºC) levam à obtenção de materiais com propriedades semelhantes, o que representa a possibilidade de um processo mais simples e curto e, consequentemente, com custos reduzidos. Em outro estudo com comprimidos obtidos a partir de géis de amido de milho ceroso retrogradado para liberação controlada da teofilina, foi observado que o processo de retrogradação levou à redução do tamanho dos poros dos géis, bem como à diminuição das propriedades de intumescimento em meio contendo enzimas. Esses efeitos contribuíram para a resistência contra erosão enzimática e o prolongamento das taxas de liberação da teofilina89. Em estudos recentes, Cardoso90 obteve com sucesso hidrogéis e micropartículas a base de misturas de goma gelana e amido retrogradado através dos métodos de geleificação ionotrópica (Al3+) e dupla reticulação (Al3+/glutaraldeído), e verificou que os sistemas apresentaram reduzidas taxas de liberação do fármaco em meio ácido e o significativo controle da liberação em pH 7,4, indicando o potencial do sistema para liberação cólon específica de fármacos. É importante ressaltar que o amido retrogradado, inicialmente explorado pela área alimentícia, também é bastante utilizado para fins nutricionais e terapêuticos, como para prevenção da Diabetes mellitus tipo 2, melhoria da saúde intestinal, saciedade, redução da glicemia91, além da diminuição dos níveis de colesterol e triglicérides92.
Amini e colaboradores93 também sugerem seu uso como um composto bioativo na prevenção do câncer colorectal, uma vez que o desenvolvimento deste tipo específico de patologia está bastante relacionado com a dieta. Estudos anteriores revelaram que o butirato (ácido graxo de cadeia curta), o principal produto de fermentação no cólon, possui atividade supressora da proliferação de células tumorais e o consumo de amido resistente eleva o conteúdo de ácido butírico.
Em face ao exposto, conclui-se que a modificação do amido pode originar materiais com diferentes propriedades físico-químicas, as quais podem se adequar a diferentes necessidades terapêuticas e levar à obtenção de sistemas de liberação prolongada de fármacos bem como propiciar a vetorização de fármacos para o cólon.
A quitina é o polissacarídeo extraído do exoesqueleto de crustáceos e resíduos da indústria de pesca, utilizada para o preparo da quitosana. As cascas secas de crustáceos possuem em torno de 15- 20% e resíduos de lula e molusco contém de 35- 40% de quitina94, constituindo um rejeito da indústria da pesca abundante e de baixo custo. A quitina também é encontrada na parede celular de fungos e outros micro-organismos, porém, o processo de extração a partir dessas fontes é de custo elevado e de difícil extração95. Após extraída, a quitina é submetida a tratamentos ácidos e básicos, os quais retiram o conteúdo mineral das cascas, promovem desproteinização e causam desacetilações brandas. Para obter a quitosana, a quitina deve ser processada em solução de hidróxido de sódio 40%, em temperatura de 120 ºC durante três horas96.
A quitina é um polímero linear, com unidades repetitivas de um dissacarídeo formado por β-(1→4) - 2-amino-2-desoxi-D-glicose e β- (1→4) -2-acetamida-2-desoxi-D-glicose, unidos por ligação glicosídica97, encontrada na natureza como um material rígido, anelástico e esbranquiçado. A estrutura química da quitina é representada na Figura 11.
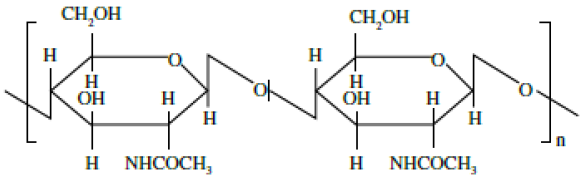
A quitosanaé originada a partir da modificação química da quitina, através de uma reação de desacetilação parcial (Figura 12), geralmente por tratamento alcalino96.
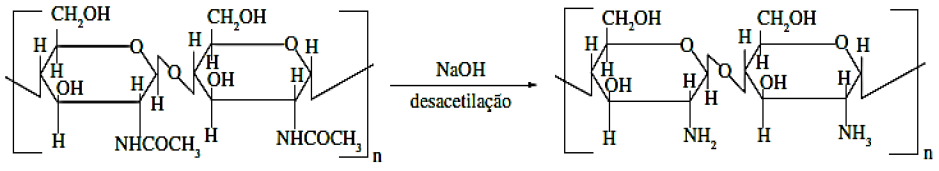
A quitosana, cuja estrutura química é representada na Figura 13, é constituída por unidades de β- (1,4)-2-amino-2-desóxi-D-glicopirano, com menores números de unidades de β-(1-4)-2- acetamido-2-desóxi-D-glicose98.
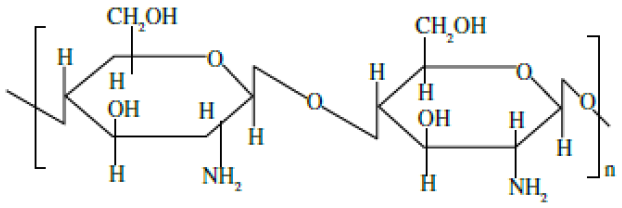
A quitosana pode ser preparada em forma de filme, permitindo sua utilização como curativo no tratamento de queimaduras. Este material é degradado por uma enzima presente na pele, a lisozima, o que anula a necessidade da retirada do mesmo, quando utilizado como curativo e por ser permeável à água e ao oxigênio, auxilia no processo de cicatrização. Além disso, a quitosana também pode ser utilizada em odontologia, na forma de gel, para diversos tipos de tratamento, inclusive para preenchimento ósseo, regeneração óssea periodontal99 e preenchimento de bolsas periodontais100.
Sua composição química, rica em aminas primárias livres leva à atividade antimicrobiana contra fungos e bactérias. Assim, a quitosana pode ser utilizada para tratar a superfície de sementes, inibindo a ação de fungos, e como cobertura comestível para frutos, além de manter a qualidade dos alimentos por evitar a perda de água e retardar o amadurecimento100. Na área farmacêutica, pode ser utilizada para liberação de fármacos para uso nasal, oral, parenteral e transdérmico99. Na área alimentícia é utilizada também em formulações para aumentar a saciedade, diminuir o colesterol e absorver a gordura ingerida na alimentação. Além das atividades descritas, a capacidade de inibição de células tumorais, ação hipocolesterolêmica e hemostática, também já foram relatadas98.
A quitosana é degrada pela enzima lisozima, pela hidrólise das ligações glicosídicas do polímero. A degradação é dependente do grau de desacetilação e cristalinidade da quitosana, sendo que quanto maior sua desacetilação, maior a degradação, porém, sua degradação é lenta, em torno de meses97.
Quanto à toxicidade, a quitosana tem se demonstrado atóxica99, e fatores de genotoxicidade não foram detectados, sendo esse um fator essencial para sua utilização em humanos, sem causar riscos à saúde96. Porém, em períodos longos de utilização como suplemento alimentar para ratos, foi detectado o bloqueio da absorção de vitaminas lipossolúveis e cálcio, acarretando retardo do crescimento, disfunções ósseas e deficiências vitamínicas. A DL50 em ratos é de 16 g/kg101.
Desenvolvida pela empresa Kelco em 1978, a goma gelana (GG) nativa é um exopolissacarídeo linear aniônico hidrofílico de elevado peso molecular sintetizado por fermentação aeróbica pelo micro-organismo Sphingomonas elodea102, também conhecido como Auromonas elodea ou Pseudomonas elodea103,107. A forma nativa é estruturalmente constituída por unidades repetidas de β-1,3-D-glicose, β-1,4-D-ácido glucurônico, β- 1,4-D-glicose e α-1,4-L-ramnose (Figura 14)106,108. Seus grupos 2-acila e 6-gliceril são removidos pela reação de hidrólise alcalina, em elevadas temperaturas, originando a GG de baixo grau de acetilação (Figura 15), comercialmente conhecida como Gelrite® ou Kelcogel®, de uso nas áreas alimentícia e farmacêutica em variados tipos de produtos102,106,109,110.
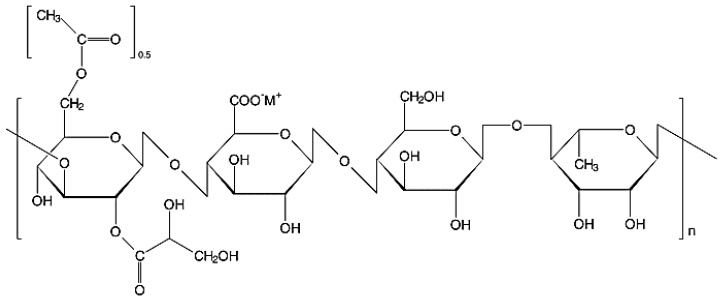
Figura 14
Estrutura química da forma nativa da goma gelana (baixa acetilação) (Adaptado de Mao, 2000)111.
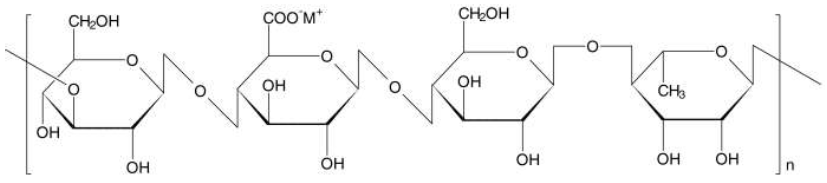
Sua característica hidrofílica e elevada capacidade de geleificação na presença de cátions di- ou trivalentes tem sido muito explorada para obtenção de novos sistemas para liberação controlada de fármacos119,120. Algumas de suas características, tais como atoxicidade, biodegradabilidade, baixo custo, propriedades mucoadesivas e degradação específica pela microbiota colônica105, a tornam um polissacarídeo atrativo e bastante promissor para o design de diferentes formas farmacêuticas.
A utilização da GG tem aumentado significativamente nos últimos anos, pois possui uma ampla aplicabilidade nas indústrias de alimentos e farmacêutica, e mais atualmente também na área da medicina regenerativa105. A gelana foi aprovada para uso alimentar em 1988 no Japão, e desperta o interesse de muitos pesquisadores devido à sua propriedade de formar géis transparentes e resistentes a variações de calor e a ambientes ácidos, mesmo quando utilizada em baixas concentrações110,120,121. A gelana utilizada como um aditivo alimentar atua como estabilizante, espessante, excelente gelificante e é capaz de produzir gel com diferentes propriedades de textura. Alguns exemplos clássicos do uso da GG em produtos alimentares são recheios para pães, confeitaria, produtos lácteos, géis sobremesa, glacês, doces e geleias, pudins, molhos, alimentos estruturados, coberturas, entre outros106.
A GG já possui uma aplicação reconhecida, de longa data, em meios microbiológicos, como alternativa ao ágar122 e como meio de crescimento bacteriano, já que seus géis termoestáveis são capazes de suportar longos períodos de incubações e elevadas temperaturas. Nessas aplicações em meios de cultura microbiológicos, características como alta pureza e a claridade do gel são vantagens adicionais que a gelana possui e que a torna um material adequado para essa finalidade106.
Na área farmacêutica, a crescente utilização da gelana pode ser exemplificada por diversos sistemas de liberação de fármacos descritos na literatura destinados ao tratamento dos mais diversos tipos de patologias, dentre eles estão os hidrogéis113, microesferas112,117, microcápsulas118,119, esferas flutuantes123, entre outros.
Recentemente, a gelana tem sido estudada para aplicações biomédicas, mais precisamente na medicina regenerativa. Suas características de biocompatibilidade e biodegradabilidade a tornam um material com grande potencial na regeneração de tecidos e seu uso para o preparo de scaffolds tem demonstrado grandes resultados124. Estudos recentes mostram que estruturas baseadas em gelana podem ser explorados como biomaterial para regeneração de cartilagens na forma de discos, membranas, fibras e partículas105,125, como scaffolds para melhorar a capacidade de regeneração óssea126 ou ainda em forma de scaffolds nanofibrosos de blendas com polivinilálcool. É utilizada como matriz para regeneração de tecidos por permitir a diferenciação e proliferação celular.
Assim, polímeros de origem natural, como a gelana, podem oferecer inúmeras vantagens, já que sua semelhança a macromoléculas biológicas, evita o reconhecimento pelo organismo como um corpo estranho127. A vantagem na utilização da GG no contexto das aplicações biomédicas fundamenta-se não só pelas suas inúmeras propriedades descritas anteriormente, mas também pela possibilidade de ser utilizada como um sistema injetável, já que possui uma similaridade estrutural com as glicosaminoglicanas presentes no tecido conjuntivo, devido à presença dos resíduos de ácido glucurônico na sua estrutura125.
A goma gelana é um dos polissacarídeos de origem natural utilizado como excipiente multifuncional para as mais variadas formas de dosagens farmacêuticas e como aditivos na indústria alimentícia. Suas propriedades particulares a tornam um material versátil, que pode ser modulado de acordo com as necessidades específicas, de modo que possa apresentar uma variedade de aplicações potenciais em áreas importantes como nos domínios da engenharia de tecidos e medicina regenerativa.
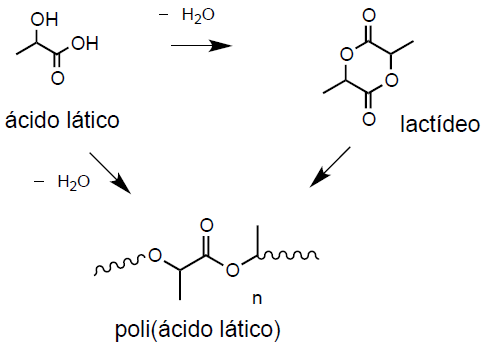
O ácido láctico é o monômero utilizado como precursor na síntese do poli (ácido láctico) (PLA). É um ácido orgânico abundante, de ocorrência natural, usado nas indústrias alimentícia, farmacêutica, cosmética, têxtil, de couro e química128. O ácido láctico (2-ácido hidroxipropiônico) é uma das moléculas quirais mais simples, e existe na forma de dois estereoisômeros, L- e D- ácido láctico, que se diferenciam pelo efeito da luz polarizada129. O ácido láctico pode ser produzido por síntese química ou fermentação.
A síntese química de ácido láctico é baseada principalmente na hidrólise de lactonitrila, derivado petroquímico, pela ação de ácidos fortes, que fornece a mistura racémica de ácido láctico D- e L-130. No caso de processos biotecnológicos, o ácido láctico pode ser facilmente sintetizado por bactérias, como por exemplo, pelos micro- organismos do gênero Lactobacillus, a partir de matéria prima de baixo custo131. Neste caso, o L- ácido láctico é preferencialmente sintetizado. O interesse na produção fermentativa de ácido láctico tem aumentado devido às perspectivas relacionadas à produção do polímero de ácido láctico, o PLA, que é totalmente biodegradável na natureza, portanto, apresenta um grande apelo ambiental. Outros dois fatores de grande importância são a vantagem da síntese por fermentação em relação à síntese química, por ser um processo de baixo impacto ambiental e alto rendimento e a utilização de produtos provenientes de fontes renováveis de baixo custo como o amido, celulose, cana de açúcar, entre outros, como nutrientes para os micro- organismos que o sintetizam por fermentação132.
O PLA é um poliéster alifático (Figura 16), de fórmula química [(C3H4O2)n]. Possui dois estereoisômeros: poli(L-ácido láctico) (PLLA) e o poli(D-ácido láctico) (PDLA), sua mistura racêmica gera o poli(D,L-ácido láctico) (PDLLA)133. O PDLA e o PLLA são imagens especulares um do outro, ambos opticamente puros e semicristalinos134, enquanto o PDLLA é racêmico, amorfo e opticamente inativo135.
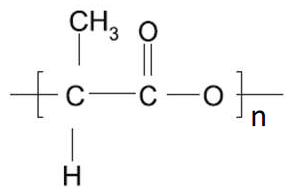
Figura 16
Estrutura química do poli(ácido láctico).
A síntese do PLA pode ocorrer por diversas rotas químicas incluindo policondensação, polimerização direta e polimerização por abertura de anel. As rotas mais utilizadas para a produção do PLA são a policondensação e a polimerização por abertura de anel136.
O processo de policondensação é realizado a vácuo, sob alta temperatura e com a utilização de solventes para a extração de água produzida pela reação de condensação. A polimerização via abertura de anel envolve um dímero cíclico de ácido láctico, o lactídeo, na presença de um catalisador. Esse processo gera um polímero de baixa massa molecular. Durante a síntese é possível associar as variáveis de tempo, temperatura e concentração do catalisador o que permite controlar as proporções e as sequências de unidades de D- e L-láctico no polímero final (Figura 17)137. A policondensação do ácido láctico resulta em um polímero de baixa massa molecular, enquanto a polimerização por abertura de anel leva a formação de polímeros de maior massa molecular138.
Devido às suas propriedades mecânicas, processabilidade termoplástica e propriedades biológicas, como biocompatibilidade e biodegradabilidade, o PLA tem se mostrado um polímero promissor139 tanto para substituição de polímeros convencionais no uso cotidiano, devido à sua degradabilidade na natureza, quanto para aplicações biológicas, devido à sua degradabilidade in vivo.
As propriedades do PLA dependem dos componentes isoméricos resultantes da quiralidade do ácido láctico, onde os dois centros assimétricos existem de quatro formas diferentes. Essas propriedades podem ser modificadas pela variação dos isômeros (L (+) / D (-)) e a homo (D (-), L (+)), da temperatura de processamento, do tempo de recozimento e da massa molecular140. O PLA pode ser amorfo ou cristalino dependendo da estereoquímica e de seu histórico térmico. O material totalmente amorfo pode ser sintetizado pelo aumento relativo do isômero D (>20%), enquanto que o material cristalino é obtido quando a quantidade de D é < que 2%. Esse polímero é relativamente rígido com temperatura de transição vítrea entre 60 ºC – 70 ºC e a temperatura de fusão entre 170 ºC- 180 ºC139.
Os poliésteres, incluindo o PLA, são polímeros hidroliticamente degradáveis. A hidrólise do PLA, através de quebras de suas ligações ésteres, gera grupos terminais carboxila e hidroxila, conforme representado na Figura 18141. Em organismos vivos, o ácido láctico gerado pela degradação do PLA é incorporado ao ciclo do ácido cítrico, resultando, ao final do processo metabólico, em subprodutos como dióxido de carbono e água142. As taxas de degradação são determinadas por fatores como o local do implante, massa molar, composição estereoquímica, cristalinidade e morfologia143.

Devido às suas propriedades, incluindo a natureza não-tóxica dos produtos de degradação, o PLA tem se mostrado atraente para a indústria biomédica, podendo ser utilizado como material de implante cirúrgico, sistema de liberação de fármacos e também como suporte poroso para o crescimento de células e tecidos com aplicação na medicina regenerativa144.
Embora o PLA se mostre um polímero promissor, algumas desvantagens como fraca ductibilidade, taxa de degradação lenta e alta hidrofobicidade, limitam suas aplicações. Desta forma estudos vem sendo realizados com a finalidade de modificar suas propriedades buscando expandir seus domínios de aplicação.
A policaprolactona (PCL) é um poliéster alifático sintetizado pela primeira vez em 1934 pelo grupo do famoso pesquisador americano Van- Natta145. Em 1958 foi relatada a primeira síntese da PCL descrevendo o mecanismo de reação através da abertura do anel do monômero pela adição de carbonato de potássio, em uma temperatura de 150 °C por 5 horas146. O processo de obtenção da PCL consiste na polimerização (por abertura do anel) do monômero de ε-caprolactona, resultando no produto de condensação de um grupo hidroxílico e um carboxílico, gerando, dentro da mesma molécula, o grupamento poliéster, pertencente à família dos poliésteres alifáticos, representado na Figura 17147. Existem pelo menos quatro tipos de mecanismos de polimerização, a polimerização aniônica, catiônica, de coordenação e radicalar. Cada método afeta algumas resultantes, como a distribuição de massa molecular, a composição do grupo terminal e da estrutura química do polímero a ser formado, e também se é em bloco, ou randomizado. Estas características, por sua vez, são importantes na definição da permeabilidade, biodegradabilidade e propriedades mecânicas do polímero148.
A PCL é um polímero hidrofóbico, semicristalino, tenaz, flexível, possui baixa temperatura de transição vítrea (Tg) entre -60 e -70ºC e funde-se a cerca de 60 ºC, apresentando boas propriedades mecânicas e grande potencial para uso como biomaterial devido à sua fácil moldabilidade em temperaturas relativamente baixas149. É solúvel em vários solventes à temperatura ambiente, incluindo tetrahidrofurano, clorofórmio, diclorometano, tetracloreto de carbono, benzeno, tolueno, cicloexanona, di-hidropirano, e 2-nitropropano. É pouco solúvel em acetona, 2- butanona, acetato de etila, acetonitrila, e dimetilformamida, e insolúvel em álcoois, éter de petróleo, e dietiléter148,150.
Esse polímero pode ser biodegradado enzimaticamente por fungos e bactérias151, porém, em animais e humanos, devido à falta de enzimas adequadas para isso, a degradação dá-se por hidrólise das ligações ésteres por ação da água, originando produtos na forma de oligômeros, ou monômeros solúveis152. O período médio de degradação do homopolímero de caprolactona in vivo é de 2 a 3 anos153,154. Quando não enzimática, a degradação desse polímero é iniciada na região amorfa no momento em que a água entra em contato com sua superfície, interagindo com os grupos carboxílicos e hidroxilas terminais, até a completa e gradativa difusão por toda a estrutura155. Esse processo hidrolisa as macromoléculas em oligômeros, e estes em monômeros, os quais se difundem para os arredores do material, caracterizando o processo de erosão. Essa erosão na superfície do material permite a entrada de uma quantidade maior de água no interior do polímero, acelerando o processo de degradação150.
A liberação rápida dos produtos de sua degradação como oligômeros e subprodutos ácidos, pode resultar em reações inflamatórias in vivo. Nestes casos, se o tecido circundante não for capaz de tamponar a alteração do pH devido à má vascularização ou baixa atividade metabólica, podem ocorrer distúrbios temporários, como, por exemplo, aumento da pressão osmótica através da acumulação de fluído locais, em casos de degradação rápida156.
O processo de biodegradação pode ser modulado de diversas formas, sendo a mais comum, a síntese de copolímeros, isto é, a associação da PCL com outros polímeros. Em geral, se associa com outras lactonas quando se quer estender o período de degradação157, ou adicionado de um DL- lactídio, por exemplo, a fim de se encurtar esse período, ou com outros polímeros para obter comportamentos específicos para a finalidade desejada148.
A PCL também tem a capacidade de formar misturas compatíveis com outros polímeros, o que pode afetar a cinética de degradação e propiciar o preparo de materiais com perfis de liberação de fármacos desejados150. Estes atributos conduzem sua aplicação no preparo de sistemas de liberação como, por exemplo, em forma de microesferas ou nanoesferas158. Diversos trabalhos relatam a pesquisa com liberação de fármacos a partir da PCL. Exemplos são a liberação de papaverina159 ou proteínas160 a partir de microesferas de PCL e a liberação de tamoxifeno a partir de nanopartículas de PCL161. Porém, uma das principais áreas de estudo ou aplicação da PCL na medicina é no desenvolvimento de scaffolds para regeneração tecidual em ossos162,163,164, cartilagens165,166,167, vasos sanguíneos168,169, peles170,172e nervos173,174. Para estas aplicações são frequentemente descritas as técnicas de copolimerização ou de modificação química da superfície do scaffold, que mantêm as propriedades do material, alterando apenas sua superfície, para melhorar a biocompatibilidade175.
O aumento das linhas de pesquisas em PCL e seus derivados reflete a versatilidade deste polímero e indica uma tendência de aumento de suas aplicações não só na área onde este é usado em substituição aos polímeros convencionais, mas também com grande potencial para as áreas médica e farmacêutica.
De forma geral, os polímeros discutidos apresentam potencial para aplicações em diversas áreas que incluem desde o preparo de novos materiais para substituição de polímeros convencionais, até materiais para aplicação médica e farmacêutica. Características como biocompatibilidade e moldabilidade são essenciais para suas aplicações nas áreas discutidas neste texto e a questão de serem, em sua maioria, provenientes de recursos renováveis é vantajosa no sentido da disponibilidade e baixo custo dos mesmos. Apesar do baixo volume desses materiais em formas disponíveis comercialmente, muitas pesquisas encontram-se atualmente focadas no desenvolvimento de produtos neles baseados, na tentativa, e com boas perspectivas, conforme indicado pelos trabalhos aqui descritos, de aumentar a gama de suas aplicações.
Figura 1
Estrutura do Fibrinogênio.
Figura 2
Polimerização e formação do polímero insolúvel de fibrina.
Figura 3
Fibrinólise.
Figura 4
Desenho esquemático da diferença entre celulose vegetal constituída de celulose, lignina e hemicelulose e a celulose bacteriana constituída pela celulose pura24.
Figura 5
.Estrutura da celulose. As linhas pontilhadas esquematizam as ligações de hidrogênio possíveis e a seta azul, a ligação.
Figura 6
Microscopia Eletrônica de Varredura da G. hansenii e a ultraestrutura da CB secretada pelo micro- organismo.
Figura 7
Representação esquemática da síntese da CB.
Figura 8
Mecanismo de reticulação da pectina de baixo grau de metoxilação na presença de íons Ca+2 73.
Figura 10
Processo esquemático da obtenção do amido retrogradado.
Figura 14
Estrutura química da forma nativa da goma gelana (baixa acetilação) (Adaptado de Mao, 2000)111.
Figura 16
Estrutura química do poli(ácido láctico).
