Estudo comparativo in vitro e avaliação da qualidade físico-química do antirretroviral aciclovir comprimidos obtidos via internet
Estudo comparativo in vitro e avaliação da qualidade físico-química do antirretroviral aciclovir comprimidos obtidos via internet
Eclética Química, vol. 41, pp. 43-53, 2016
Universidade Estadual Paulista Júlio de Mesquita Filho

Este trabalho está sob uma Licença Internacional Creative Commons Atribuição 4.0.
Resumo: O aciclovir é um fármaco análogo do nucleosídeo da guanosina que inibe a replicação viral através da interrupção da cadeia de DNA com alta especificidade para os vírus do herpes. É necessário que consumidores de medicamentos antirretrovirais obtenham produtos confiáveis do mercado farmacêutico, e para isso, os ensaios analíticos contribuem para comprovar a sua qualidade durante o seu prazo de validade. Este trabalho tem por objetivo realizar um estudo comparativo in vitro e avaliar a qualidade de comprimidos de aciclovir 200 mg disponíveis no mercado virtual. Os métodos utilizados para realizar os testes de controle de qualidade físico- química de aspecto, doseamento, dissolução, desintegração, dureza, friabilidade, peso médio, umidade e uniformidade, seguiram as recomendações das literaturas. Os resultados obtidos indicaram que as especialidades farmacêuticas cumpriram com seus parâmetros de qualidade, porém, apresentaram resultados de friabilidade, uniformidade e de teor de umidade próximos aos limites máximos preconizados pela Farmacopeia Brasileira, 5ª Ed. Diante disso, a avaliação da qualidade durante o prazo de validade dos produtos obtidos pela internet tem implicações na segurança e eficácia dos medicamentos, e isso deve ser considerado pelos profissionais de saúde para salvaguardar a terapêutica dos pacientes.
Palavras-chave: Aciclovir, avaliação da qualidade, mercado virtual.
Abstract: Acyclovir is an analog of guanosine nucleoside drug which inhibits viral replication by interrupting DNA chain with high specificity for the herpes virus. It is necessary that consumers of antiretroviral drugs obtain reliable products from pharmaceutical market, and for this, the analytical tests contribute to prove its quality during its shelf life. This work aims to carry out a comparative study in vitro and evaluate the quality of acyclovir 200 mg tablets available in the virtual market. The methods used to perform the physical-chemical quality control testing aspect, assay, dissolution, disintegration, hardness, friability, weight, humidity and uniformity followed the recommendations of the literature. The results indicated that the medicinal products have met with their quality parameters, but presented results of friability, uniformity and moisture content near the maximum limits recommended by the Brazilian Pharmacopoeia, 5th Ed. Therefore, the quality assessment over the shelf life of the products obtained via internet has implications for the safety and efficacy of the drugs, and this should be considered by health professionals to safeguard the treatment of patients.
Keywords: Acyclovir, quality assessment, virtual market.
INTRODUÇÃO
O sucesso do tratamento antirretroviral veio com a descoberta do aciclovir (ACV), um fármaco análogo do nucleosídeo da guanosina com alta especificidade para os vírus do herpes. Sua força de atuação varia conforme o vírus e é mais ativo contra o vírus do herpes simples e menos ativo contra o vírus varicela- zoster1,2.
Quando administrado por via oral, o medicamento com dosagens de 200 e 400 mg tem sua biodisponibilidade entre 10% a 30%, não sendo proporcional ao aumento da dose. Sua distribuição é ampla pelos líquidos corporais com tempo de meia- vida de 2,5 horas. Seu metabolismo é feito pelas enzimas hepáticas do citocromo P450 e sua eliminação dá-se por excreção renal, principalmente por secreção tubular1.
Em razão de o Brasil ser conhecido como um dos maiores vendedores virtuais e também produtores de medicamentos do mundo, é observado que o crescimento no consumo de medicamentos aumentou de forma significativa. Diante disso, necessita-se de produtos confiáveis no mercado farmacêutico e de realização de ensaios analíticos que comprovem sua qualidade durante seu prazo de validade3,6,10.
A qualidade de um produto é considerada como a soma de características e propriedades satisfatórias, a fim de atender as necessidades dos consumidores, com isso o controle da qualidade dos medicamentos na indústria farmacêutica é indispensável para sua comercialização. A qualidade de um medicamento é avaliada em diversas etapas, inicia-se na análise das matérias-primas, ocorre durante o processo produtivo, na obtenção do produto acabado e na sua comercialização4,5,6.
Muitos medicamentos são adquiridos via internet e transportados pelo sistema de transporte de encomendas para diferentes regiões do país. O processo de transporte inadequado pode ocasionar alteração na qualidade do produto que será entregue ao consumidor. No transporte das formas farmacêuticas pelo sistema de transporte de encomendas, esses produtos podem sofrer intensas variações climáticas e físicas, como alterações na temperatura, exposição à luz, umidade, fricção e atrito. Essas variações contribuem para reduzir a qualidade do produto farmacêutico que será entregue ao consumidor3,6,10.
É indispensável realizar estudos comparativos entre medicamentos genéricos e referência, uma vez que ensaios analíticos in vitro são determinantes para provar a semelhança entre os produtos7. Realizar testes de controle de qualidade físico-química em medicamentos avalia se a forma farmacêutica atende as especificações estabelecidas pela Farmacopeia Brasileira ou por outros compêndios oficiais. Se durante o estudo houver a reprovação em algum ensaio, é comprovado o comprometimento da qualidade do produto, o que comprometerá a segurança e a eficácia do medicamento6,8.
Diante do exposto, este trabalho tem por finalidade realizar ensaios comparativos in vitro e avaliar a qualidade físico-química de comprimidos dos medicamentos referência e genérico obtidos via internet, que contêm 200 mg do antirretroviral ACV, por meio dos ensaios de aspecto, doseamento, dissolução, desintegração, dureza, espessura, diâmetro, friabilidade, peso médio, umidade e uniformidade.
MATERIAL E MÉTODOS
Amostras, substância química de referência e reagentes
Comprimidos de duas diferentes especialidades farmacêuticas denominados de A (medicamento referência) e B (medicamento genérico), contendo 200 mg de ACV, foram adquiridos via internet de drogarias localizadas na cidade de São Paulo – SP e transportados para a UFMT/CUA – Barra do Garças – MT. Foi utilizado como substância química de referência ACV (SQR) produzido pela Farmacopeia Brasileira, lote: 1021. Todos os reagentes empregados apresentaram grau analítico.
Equipamentos
Balança analisadora de umidade por infravermelho (Gehaka, mod. IV2000), balança analítica (Celtac, mod. FA2104N), desintegrador (Nova Ética, mod. 301AC/03), dissolutor (Nova Ética, mod. 299 - 6TS), durômetro manual (Nova Ética, mod. 298/DGP), espectrofotômetro (Biospectro, mod. SP - 220), friabilômetro (Nova Ética, mod. 300/1), paquímetro 6” (Zaas precision), sistema de purificação de água Milli-Q-plus (Millipore) e ultrassom (Unique, mod. USC – 2800).
Avaliação da qualidade dos comprimidos de aciclovir 200 mg
Este estudo comparativo in vitro que avalia a qualidade de comprimidos de ACV 200 mg, obtidos via internet, seguiu os parâmetros estabelecidos pela Farmacopeia Brasileira, 5ª edição (volumes I e II) e literaturas1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28.
Aspecto dos comprimidos
Foram analisados 10 (dez) comprimidos de forma aleatória, a fim de observar as seguintes características: cor, odor, presença ou ausência de logomarca, sulco e revestimento, assim como formato e integridade das especialidades farmacêuticas9,10,11.
Doseamento de aciclovir
Foram pesados 20 (vinte) comprimidos e em seguida pulverizados com auxílio de gral e pistilo. Foi transferido 0,1 g de ACV para balão volumétrico de 100,0 mL e adicionado 60 mL de hidróxido de sódio (NaOH) 0,1 mol L-1 . Após 15 minutos em ultrassom, o volume do balão volumétrico foi completado com o mesmo diluente (NaOH 0,1 mol L-1 ), homogeneizado e filtrado. Foram transferidos 15,0 mL da solução obtida para balão volumétrico de 100,0 mL, adicionou-se 50 mL de água purificada obtida por osmose reversa, 5,8 mL de ácido clorídrico (HCl) 2,0 mol L-1 , foi completado o volume com água purificada obtida por osmose reversa e em seguida homogeneizou-se a solução. Em seguida, 5,0 mL foram transferidos para balão volumétrico de 50,0 mL e o volume foi completado com ácido acético 0,1 mol L-1 e homogeneizado. Foi preparada uma solução contendo a SQR de ACV na concentração de 0,0015%, utilizando os mesmos solventes. As amostras foram preparadas em triplicata e as leituras das absorbâncias das soluções resultantes foram medidas em cubetas de quartzo de 1 cm à temperatura ambiente (27 ± 1) ºC , no comprimento de onda de 255 nm. Empregou-se HCl 0,1 mol L-1 para ajuste do zero12.
O teor foi calculado com a Equação 1:
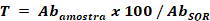 [Equação 1]
[Equação 1]Em que, T = Teor; Abamostra = leitura de absorbância obtida pela solução amostra; AbSQR = leitura de absorbância obtida pela solução que contém a substância química de referência de ACV10,12,15.
Teste de dissolução
Para o teste de dissolução foi utilizado o equipamento dissolutor. Foram analisados 6 comprimidos de ACV de cada especialidade farmacêutica12.
O meio de dissolução utilizado foi HCl 0,1 mol L-1 (900 mL) a uma temperatura de (37 ± 0,5) ºC, sob rotação de 50 rpm/min. Após 45 minutos, foram retiradas alíquotas do meio de dissolução, que foram diluídas, quando necessário, com ácido clorídrico 0,1 mol L-1, até concentração de 0,01 mg/mL. As leituras das absorbâncias foram medidas em 255 nm a temperatura ambiente (27 ± 1) ºC e foi utilizado o mesmo solvente para ajuste do zero. Foi calculada a quantidade de ACV dissolvido no meio, a partir da comparação das leituras obtidas para a solução que continha a substância química de referência, na concentração de 0,001% (m/V). A SQR foi solubilizada previamente em ácido clorídrico 0,1 mol L-1 10,12,13. As amostras foram preparadas em duplicata.
A quantidade de ACV dissolvida foi calculada conforme demonstrado na Equação 2. mol L-1, foi completado o volume com água purificada obtida por osmose reversa e em seguida homogeneizou-se a solução. Em seguida, 5,0 mL foram transferidos para balão volumétrico de 50,0 mL e o volume foi completado com ácido acético 0,1 mol
 [Equação 2]
[Equação 2]Em que, Qdissolvido = Quantidade dissolvida de ACV dissolvido em HCL 0,1 mol L-1 em 45 min; Abamostra = leitura de absorbância obtida pela solução amostra; AbSQR = leitura de absorbância obtida pela solução que contém a SQR10,12,13.
De acordo com a Farmacopeia Brasileira, 5ª Ed., não menos que 80 + 5% (Qdissolvido) da quantidade declarada de ACV devem se dissolver em 45 minutos para ser aprovado em primeiro estágio (E1)12.
Tempo de desintegração
Para a realização deste ensaio foi necessária a utilização de aparelhagem específica, o desintegrador. Este teste consiste na utilização de 6 comprimidos, colocados unitariamente em 6 tubos de uma cesta com fundo metálico, e sobre os comprimidos foi adicionado um disco de acrílico. Após esse procedimento, a cesta foi submetida a movimentos suaves e verticais em solução aquosa a temperatura de (37 ± 1) ºC durante 30 minutos. Ao final desse intervalo, foi avaliado o estado físico de cada comprimido8,10,14.
Cumprem o teste os comprimidos que não deixarem resíduos na tela metálica do aparelho, ou comprimidos que, quando transformados em pasta, apresentarem núcleo seco após ser palpável no tempo final de desintegração10,14,15.
Teste de dureza, espessura e diâmetro
A determinação da dureza foi realizada no aparelho denominado durômetro. O ensaio foi procedido conforme as recomendações da Farmacopeia Brasileira 5ª Ed., que preconiza a utilização de 10 comprimidos para que, individualmente, seja medida a força necessária para esmagá-los diametralmente, medida em Newton (N). A espessura e o diâmetro desses comprimidos foram medidos de forma aleatória, em 10 (dez) unidades, e com a utilização de um paquímetro. Os resultados encontrados são de caráter informativo8,14.
Teste de Friabilidade
Para realizar a friabilidade (Fr) foram pesados 20 (vinte) comprimidos de ACV 200 mg e em seguida as unidades foram transferidas para um aparelho denominado friabilômetro. Foram ajustadas as rotações por minuto e o tempo, de modo que ocorressem 100 rotações durante 4 minutos. Após o teste, os comprimidos foram limpos e pesados novamente8,12.
A Fr é representada pela diferença entre a massa inicial (Mi) e a massa final (Mf) dos comprimidos, de acordo com a Equação 3.
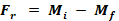 [Equação 3]
[Equação 3]Em que Fr = Friabilidade; Mi = massa inicial dos comprimidos e Mf = massa final dos comprimidos.
O cálculo para obtenção da friabilidade em função da porcentagem de pó perdido (Pp%) é determinado conforme Equação 4.
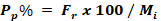 [Equação 4]
[Equação 4]Em que, Pp% = Porcentagem de pó perdido durante o teste de friabilidade; Fr = Friabilidade e Mi = massa inicial.
Para o lote ser considerado aprovado, o resultado obtido de Pp% não deverá ultrapassar 1,5% do Mi e ao finalizar o teste nenhum comprimido poderá ser encontrado lascado, quebrado, rachado ou partido14,16.
Determinação de peso médio
Neste ensaio foi determinada a massa separadamente de 20 (vinte) comprimidos em balança analítica. Em seguida foi avaliada a variação percentual das massas das unidades em relação à massa média, conforme Equação 5.
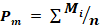 [Equação 5]
[Equação 5]Em que Pm = peso médio; Ʃ Mi = Soma das massas dos valores individuais e n = número de comprimidos avaliados10,14:
Foram calculados os limites de variação máximo e mínimo, segundo a Equação 6.
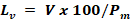 [Equação 6]
[Equação 6]Em que Lv = limite de variação; V = Massa unitária máxima e mínima encontrado no teste de peso médio e Pm = peso médio.
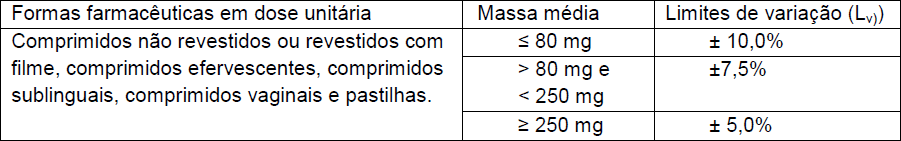
Cumprem o teste os comprimidos que, preferencialmente, não ultrapassarem os limites superiores máximos e mínimos, conforme Quadro 1. Caso a massa individual esteja fora do especificado, tolera-se não mais que duas unidades e nenhuma unidade acima ou abaixo do dobro das porcentagens indicadas (Quadro 1).
Teste de verificação de umidade
Foi determinada na balança analisadora de umidade por infravermelho a porcentagem de água em 1,0 g de pó de comprimidos, previamente pulverizados no ensaio de doseamento. A amostra foi mantida no equipamento a uma temperatura de 105º C por 5 min, em seguida foi obtida a quantidade de água presente no pool analisado 17,18. A umidade não deve ultrapassar 6,0%, conforme especificado pela monografia do produto14.
Teste de uniformidade
Para a determinação da uniformidade foi utilizado o método de variação de peso14. Neste parâmetro é recomendado separar 30 unidades de cada especialidade farmacêutica e retirar, de maneira aleatória, 10 unidades para análise.
Os resultados individuais de uniformidade por peso deverão ser expressos em porcentagem da quantidade declarada pela Equação 7.
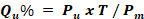 [Equação 7]
[Equação 7]Em que Qu% = quantidade de fármaco em cada unidade; Pu = peso unitário dos comprimidos; T = Teor obtido no teste de doseamento e Pm = peso médio.
Para avaliação dos resultados de uniformidade por peso, foi calculado o valor de aceitação (VA) de acordo com as equações descritas no Quadro 2. Cumpre o teste de uniformidade VA inferior a 1512,14.
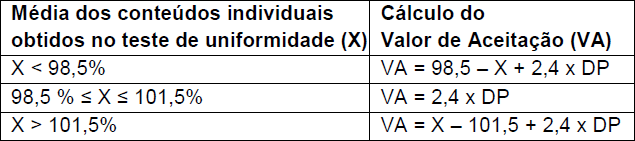
X = média dos conteúdos individuais obtidos no teste de uniformidade; VA = valor de aceitação e DP: desvio padrão.
RESULTADOS E DISCUSSÃO
Avaliar a qualidade de especialidades farmacêuticas durante o seu prazo de validade possibilita verificar se a forma farmacêutica produzida e dispensada à população cumpre com os critérios determinados na monografia do produto durante o seu período de comercialização14,19.
A verificação do aspecto permitiu avaliar o formato da forma farmacêutica, a presença de sulco, assim como a coloração das unidades dos comprimidos. É preconizado na literatura que o medicamento genérico seja equivalente farmacêutico do medicamento referência, podendo apresentar aspectos distintos, desde que estes não alterem a biodisponibilidade do fármaco14,19.
Cor e formato são características que variam de um laboratório para o outro, pois são métodos adotados para facilitar o processo de produção, identificação do produto ou mesmo melhorar a estética dos comprimidos14,19.
Foi observado que os comprimidos em análise apresentavam aspectos diferentes. Os medicamentos avaliados dispunham de aspecto uniforme e mantiveram a sua integridade após a blistagem. Os comprimidos A possuíam coloração branca, formato hexagonal e plano, com presença de logomarca, ausência de odor, sulco e revestimento. Já os comprimidos B apresentavam cor azul, formato circular e abaulado, sem odor, ausência de sulco, logomarca e revestimento.
A administração de um medicamento com concentração de fármaco inadequada acarreta em falha terapêutica e uma alta dosagem poderá ocasionar intoxicação, de qualquer forma, poderá comprometer a saúde do paciente. Diante disso, é de extrema importância que o medicamento cumpra com as suas especificações de qualidade, durante e após o processo de produção, até o término do seu prazo de validade14. A monografia de ACV comprimidos da Farmacopeia Brasileira, 5 ª Ed., especifica que o teste de doseamento deverá apresentar no mínimo 95,0 % e no máximo 105,0% de fármaco em relação à quantidade declarada. Os comprimidos A e B apresentaram teores de 101,9% e 98,5% respectivamente. Ambos os produtos testados estão em conformidade com a especificação farmacopeica.
Ao se comparar os resultados obtidos neste ensaio é observado que os produtos analisados apresentaram valores de doseamento próximos, com uma diferença de dosagem menor que 2,0%. Isso indica que a quantidade de fármaco disponível em ambas as especialidades farmacêuticas não possui diferença significativa10,14.
É verificada a importância da realização do teste de doseamento, porque por meio desse ensaio é possível determinar a quantidade de fármaco presente nos comprimidos e avaliar se a especialidade farmacêutica cumpre com esse parâmetro de qualidade, uma vez que é possível encontrar no mercado farmacêutico medicamentos com critérios fora dos padrões estabelecidos para consumo14,20.
O ensaio de dissolução possibilita a avaliação da liberação do fármaco pelas partículas desintegradas, sendo esse um dos ensaios mais relevantes para verificar a qualidade de um medicamento, uma vez que o processo de dissolução tem relação direta com a eficácia do produto nos ensaios “in vivo”. Baseando- se nessas informações, existe uma grande preocupação para que o produto esteja dentro dos padrões especificados na literatura13,14,21.
Foi avaliada, nas especialidades farmacêuticas A e B em estudo, a quantidade de ACV dissolvido no meio de dissolução após tempo e rotação preconizados pela Farmacopeia Brasileira, 5ª Ed. (45 minutos a 50 rpm). A quantidade de fármaco dissolvida em cada cuba está representada na Tabela 1.
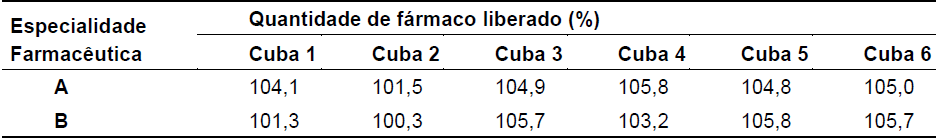
É especificado na literatura que serão aprovados em E1 medicamentos que apresentarem resultados maior ou igual a Q + 5% em todas as unidades testadas, sendo Q a quantidade do fármaco dissolvido indicada em sua monografia14. Conforme demonstrado na Tabela 1, todas as cubas analisadas estão aprovadas, porque apresentaram valores de liberação de ACV acima de 80,0 + 5%.
Ao avaliar os resultados dos ensaios de dissolução de aciclovir para as formulações A e B é observado que os valores encontrados neste teste são maiores aos obtidos em doseamento. Muitos fatores são responsáveis por favorecer a solubilização de fármacos, entre eles podemos destacar a temperatura. Ao avaliar a técnica estabelecida pela Farmacopeia Brasileira, 5ª Ed., para análise de doseamento de aciclovir, o método não estabelece a utilização de temperatura, o que pode reduzir a solubilização do fármaco no ensaio de teor. Em 2015, Azeem e colaboradores realizaram um estudo de dissolução e algumas amostras deste ensaio apresentaram liberação in vitro de fármaco superior aos resultados de doseamento, isso também é observado no estudo de Laporta e colaboradores em 201329,30.
A desintegração de comprimidos pode influenciar na atividade terapêutica, pois quando o comprimido não tem uma desintegração adequada, isso poderá reduzir a dissolução e absorção do fármaco. Diante disso, a desintegração deve ocorrer de forma apropriada, transformando o comprimido em partículas menores, o que favorecerá a dissolução, absorção e biodisponibilidade do fármaco no organismo6,15,21.
As amostras A e B desintegraram em tempo inferior a 30 minutos, que é o limite máximo preconizado para desintegração de comprimidos de liberação rápida sem revestimento12. As especialidades A e B desintegraram em 60 e 50 segundos respectivamente. A partir dos resultados é observado que os produtos submetidos ao teste estão em conformidade com o especificado, e não apresentaram diferenças significativas no tempo de desintegração.
Ao comparar o tempo máximo para desagregação dos comprimidos e os tempos atingidos nos ensaios, é verificado que ambos os produtos apresentaram uma desintegração relativamente rápida. Essa rápida velocidade de desintegração pode ter sido influenciada por fatores como presença de superdesintegrantes nas formulações dos medicamentos, força de compressão, e até mesmo mau acondicionamento e transporte do produto6,22.
Em 2004, Gennaro e colaboradores relataram que a agitação turbulenta na realização do teste de desintegração não condiz com o que ocorre no processo in vivo, e por isso, não é um bom indicador para avaliar a liberação do fármaco, não podendo relacionar o baixo tempo de desintegração com maior eficácia do medicamento.
A dureza dos comprimidos determina a sua resistência mecânica. Os comprimidos devem resistir aos atritos sofridos durante o processo de blistagem, quando são armazenados, transportados e também no manuseio antes da administração pelo usuário. Embora um elevado grau de dureza traga os benefícios citados acima, se esta for alta, poderá influenciar na desintegração e dissolução dos comprimidos, o que comprometerá a biodisponibilidade do fármaco21.
Os comprimidos em estudo, A e B, apresentaram valores médios de resistência 82 N ± 13% e 70 N ± 8% respectivamente (Tabela 2). A literatura não preconiza valores mínimos ou máximos para o ensaio de dureza, pois os caracteriza apenas como resultados informativos12.
Ao avaliar os comprimidos em suas embalagens primárias foi possível observar que não houve quebra ou ruptura de nenhuma unidade, isso também ocorreu após a retirada dos comprimidos do blíster. Estas características indicaram que as amostras apresentaram dureza adequada, pois quanto maior o seu valor, menor a chance dos comprimidos sofrerem rupturas e/ou desgastes8,14.
Ao confrontar os resultados de dureza deste estudo com o ensaio de dissolução, é observado que o teste que avalia a resistência mecânica não influenciou de forma significativa na liberação do fármaco, pois ambas as especialidades farmacêuticas atingiram valores médios de liberação de ACV próximos a 100,0% no tempo de dissolução (45 minutos) (Tabela 1)12,14. Para ensaios in vitro de formas farmacêuticas de liberação imediata são determinados valores de dissolução média de no mínimo 75,0% da substância ativa em até 45 minutos24.
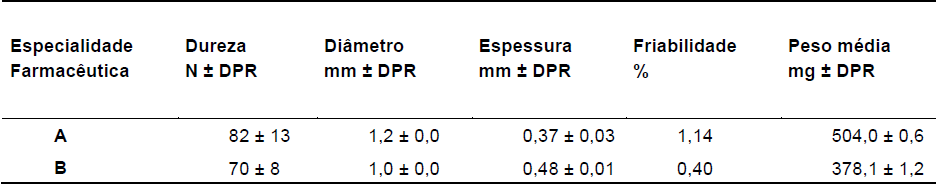
As amostras de ACV A e B foram dimensionadas e apresentaram, respectivamente, valores de diâmetro de (1,2 ± 0,0) mm e (1,0 ± 0,0) mm e de espessura (0,37 ± 0,03) mm e (0,48 ± 0,01) mm (Tabela 2).
Após análise dos resultados foi verificada uniformidade no formato das amostras de cada produto analisado, pois alterações significativas nos valores de espessura e diâmetro indicam possíveis desvios de peso nos comprimidos e nos resultados de dureza, o que poderia comprometer a qualidade do produto25.
É determinado que o valor de friabilidade seja inferior a 1,5% e que nenhum comprimido apresente no final do teste danos como quebra ou rachadura. Estas condições indicam que os produtos serão resistentes aos impactos da manipulação e do transporte10,12. Os resultados descritos na Tabela 2 demonstram que o medicamento A apresentou um valor de friabilidade maior (1,14%), quando comparado com B (0,40%). Isso ocorreu em função do formato hexagonal do comprimido A, pois ao ser submetido ao teste suas bordas foram desgastadas pelo impacto, o que possibilitou aumentar a perda de pó da forma farmacêutica.
Após comparar neste estudo os resultados encontrados nos testes de resistência mecânica dos produtos adquiridos pela internet, foi possível observar que o transporte dos antirretrovirais, da cidade de São Paulo – SP para a cidade de Barra do Garças – MT, não influenciou de maneira significativa nos valores de dureza e friabilidade, pois as amostras A e B cumpriram com suas especificações estabelecidas pela Farmacopeia Brasileira, 5ª Ed. (Tabela 2).
O ensaio de peso médio é uma ferramenta importante para o controle de qualidade, pois se não houver coerência e uniformidade das massas individuais dos comprimidos com os parâmetros estabelecidos, o lote do produto deve ser reprovado14,22.
Em processos produtivos há uma dificuldade em manter constante a uniformidade de massa nas unidades produzidas, e é preferível a menor variação possível. Pela literatura essa variação é aceita, desde que não ultrapasse os valores previamente estipulados pela monografia do produto, e quanto menor o grau de variação, melhor a sua uniformidade em relação à massa14,22.
Ao utilizar a ferramenta da qualidade gráfico controle é verificada a variação das massas individuais em ambos os produtos nota-se que nenhuma amostra ultrapassou os limites inferior (LI) (A = 479 mg; B = 359 mg) e superior (LS) (A = 529 mg; B = 397 mg) (Figura 1), e a maioria dos comprimidos apresentou massas próximas à massa média (A = 504,0 ± 0,6%; B = 378,1 ± 1,2%) (Tabela 2). Isso é observado principalmente para os comprimidos do medicamento A, o que indica que há um maior controle da massa durante a sua produção em relação ao medicamento B, pois o DPR encontrado em A (DPR = 0,61%) é menor quando comparado com B (DPR = 1,25%) (Tabela 2).
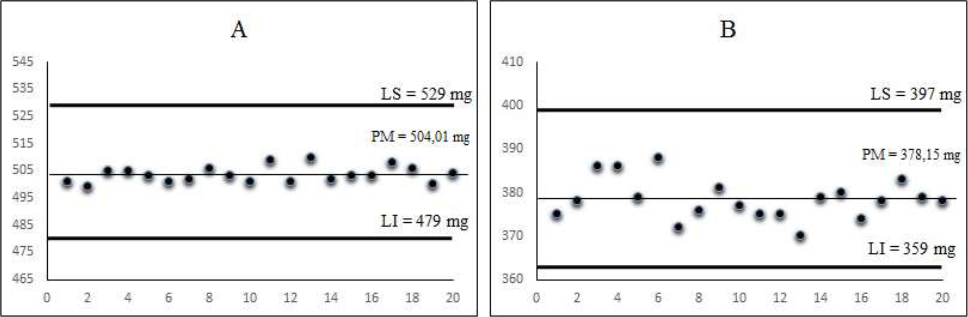
Figura 1
Gráfico de controle dos medicamentos A e B que avalia a massa individual de comprimidos de aciclovir 200 mg no ensaio de peso médio.
Para comprimidos com massa média acima de 250 mg é estabelecida uma variação entre as unidades de no máximo ± 5,0% e é tolerável não mais que duas e nenhuma unidade acima ou abaixo do dobro das porcentagens indicadas12,14. Neste estudo, todas as amostras analisadas não ultrapassaram os LI e LS (Figura 1).
É importante a avaliação e determinação da umidade em formas farmacêuticas sólidas, já que uma quantidade elevada de água influencia na contaminação microbiológica, favorece reações de oxidação ou hidrólise no fármaco e excipientes, formação de possíveis produtos de degradação tóxicos, o que poderá alterar a estabilidade e a ação terapêutica do medicamento27.
Após a realização do teste de umidade foram encontrados valores de 5,2% e 5,0% para os medicamentos A e B respectivamente. As amostras apresentaram resultados próximos ao limite máximo especificado pela Farmacopeia Brasileira, 5ª Ed. (6,0%). Esses altos índices de umidade nas especialidades farmacêuticas deste estudo podem comprometer a estabilidade do medicamento e possibilitar a formação de produtos de degradação e a contaminação microbiológica. Esses fatores poderão ocasionar alterações no prazo de validade do produto acabado, comprometendo a qualidade do medicamento que está disponível no mercado nacional e virtual3,27. O transporte inadequado das formas farmacêuticas também pode ocasionar alteração na qualidade do produto que será entregue ao consumidor. Pelo sistema de transporte de encomendas, esses produtos podem sofrer intensas variações climáticas e físicas, como alterações na temperatura, exposição à luz, umidade, fricção e atrito. Estas variações contribuem para reduzir a qualidade do medicamento6,10.
O teste de uniformidade determina a quantidade de fármaco presente em cada conteúdo avaliado. Nos lotes de medicamentos que cumprem com o especificado para esse teste, é observado que o processo de produção foi adequado e favoreceu a distribuição homogênea do fármaco nas unidades avaliadas7,14,21.
Foi avaliada a uniformidade por massa de 10 unidades de cada especialidade farmacêutica de antirretrovirais adquiridos via internet e encontraram- se médias de 101,3% (Desvio padrão (DP) = 2,52) e 98,77% (DP = 5,40%); com VA de 6,05 e 12,96 para as amostras A e B respectivamente. Os VA foram calculados pela equação estabelecida pela Farmacopeia Brasileira, 5ª Ed.; VA = 2,4 x DP; porque a média dos conteúdos individuais (X) obtidos no teste de uniformidade para ACV pelo teste de doseamento está entre 98,5% ≤ X ≤ 101,5%. As amostras analisadas cumprem com o teste de uniformidade, pois apresentaram VA inferior ao estabelecido pela literatura (VA ≤ 15)12.
É importante avaliar a uniformidade das unidades dos medicamentos comercializados, uma vez que é necessário que a quantidade de fármaco esteja o mais próximo do valor declarado, porque isso irá garantir uma administração com dose adequada e será obtida uma melhor ação terapêutica ao paciente28.
CONCLUSÕES
Consumidores que pretendem utilizar aciclovir no tratamento podem obtê-lo pela internet e com pouca dificuldade. Os resultados encontrados neste trabalho indicaram que as especialidades farmacêuticas cumpriram com seus parâmetros de qualidade, porém, apresentaram resultados de friabilidade para A (1,14%), uniformidade para B (VA = 12,96) e umidade para A e B (5,2% e 5,0% respectivamente) próximos aos limites máximos estabelecidos pela Farmacopeia Brasileira, 5ª Ed. Diante disso, a avaliação físico-química durante o prazo de validade das formas farmacêuticas tem implicações para a qualidade e segurança dos medicamentos, e isso deve ser considerado pelos profissionais de saúde para salvaguardar a terapêutica dos pacientes e inspecionar os produtos disponíveis no mercado farmacêutico.
Gratidão
Ao Laboratório de Análise e Produção Farmacêutica (LAPPHARMA) da Universidade Federal de Mato Grosso – Câmpus Universitário Araguaia.
REFERÊNCIAS
1. L. L. Brunton, B. A .Chabner, B. C. Knollmann, As bases farmacológicas da terapêutica de Goodman & Gilman, AMGH Editora Ltda, Porto Alegre, 12ª ed.:, (2012).
2. H. P. Rang, M. M. Dale, J. M. Ritter, R. J. Flower, Farmacologia, Elsevier, Rio de Janeiro, 6ª ed., (2007).
3. V. Aida, M. Beatriz, A. Gisela, Perigo on-line, Revista Época, 2005, p. 98.
4. L. F. Kohler, H. D. Nascimento, E. L. L. Schwengber, Z. M. P. Bandeira, G. V. Pazin, S. R. P. Machado, Avaliação biofarmacotécnica e perfil de dissolução de comprimidos de dipirona: equivalência farmacêutica entre medicamentos de referência, genéricos e similares, Rev. Bras. Farm., 2009, 90, 309-315.
5. C. F. A. Camargo, V. B. Sa, L. G. Nogueira, Estudo comparativo de dipirona gotas entre medicamentos de referência, genérico e similar comercializados na cidade de Trindade [Monografia], Trindade, GO: Faculdade União de Goyazes, (2011).
6. L. H. C. Vaz, Estudo comparativo de equivalência farmacêutica e perfil de dissolução “in vitro” do paracetamol fabricado no Brasil e na Holanda [Monografia], Universidade do Extremo Sul Catarinense, Criciúma, SC, (2013).
7. C. Rigobello, A.V. Gasparetto, A. Diniz, M.F. Rabito, M.M.F. Nery,. Avaliação da qualidade e perfil de dissolução de comprimidos de cloridrato de propranolol, Acta Sci., 2013, 35, 85-90.
8. M. A. Lamolha, A. C. P. Rodrigues, B. C. Silva, F. C. Granata, G. S. Podavin, J. C. O. Lima, Avaliação da equivalência farmacêutica de furosemida em comprimidos de 40 mg, Rev. Bras. Farm., 2012, 93, 17-21.
9. H. G. Ferraz, Avaliação biofarmacêutica “in vitro” e “in vivo” (bioequivalência) de comprimidos de ampicilina 500 mg comercializados no Brasil [Tese], São Paulo, SP: Universidade de São Paulo, (1997).
10. V. L. Castro, Estudos de equivalência farmacêutica com comprimidos do cloridrato de bupropiona em medicamentos similar e de referência [Dissertação], Goiânia, GO, Pontífica Universidade Católica de Goiás, (2010).
11. J. J. R. G. Pinho, S. Storpirtis, Estudo comparativo “in vitro” das propriedades biofarmacotécnicas de comprimidos de cloridrato de metformina comercializados no Brasil, Rev. Bras. Cienc. Farm., 2001, 37, 95-105.
12. Farmacopeia Brasileira, Parte II, São Paulo: Atheneu, 5ª ed.,(2010).
13. M. C. Alves, H. C. Polonini, U. P. Vaz, A. O. Ferreira, M. A. F. Brandão, Estudo comparativo de preparações orais sólidas contendo carbamazepina: teste e perfil de dissolução, Rev. Bras. Farm., 2012, 93, 487-492.
14. Farmacopeia Brasileira, Parte I, São Paulo: Atheneu, 5ª ed., (2010).
15. V. C. B. Oliveira, R. Campos, Estudos de equivalência farmacêutica de comprimidos de ibuprofeno [Monografia]. Curitiba (PR) : UniBrasil, (2014).
16. L. M. Linsbinski, C. R. Musis, S. R. P. Machado, Avaliação da equivalência farmacêutica de comprimidos de captopril, Rev. Bras. Farm., 2008, 89, 214-219.
17. D. B. Borges, M. R. Farias, E. P. Simões, E. P. Schenkel, Comparação das metodologias da Farmacopeia Brasileira para determinação de água em matérias-primas vegetais, e validação da determinação de água em analisador de umidade para Calendula officinalis L., Foeniculum vulgare Miller, Maytenus ilicifolia Mart. ex. Reissek e Passiflora alata Curtis, Rev. Bras. Farmacogn., 2005, 15, 229-236.
18. D. S. Silva, Comparação de métodos de determinação de umidade em matérias-primas de uso farmacêutico [Monografia]. Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, (2010).
19. L.V. Allen Jr., N. G. Popovich, H. C. Ansel, Formas Farmacêuticas e Sistemas de Liberação de fármacos. Porto Alegre, Editora Artmed, 8 ed., (2007).
20. M. D. Bianchin, C. R. Blatt, A. S. Soares, I. C. Külkamp-Guerreiro, Avaliação da qualidade de comprimidos de propranolol e enalapril distribuídos no sistema público de saúde em uma cidade do sul do Brasil, Ciên. Saúde Colet., 2012, 17, 491-498.
21. A. C. Rocha, Análise da qualidade físico-química de comprimidos de cloridrato de propranolol dispensados pelo programa Farmácia Popular do Brasil [Monografia]. Rio de Janeiro, RJ : Instituto Federal de Educação, Ciência e Tecnologia do Rio de Janeiro, (2013).
22. C. S. Bueno, D. Weber, A. C. Moreira, Avaliação da qualidade de quatro especialidades farmacêuticas contendo hidroclorotiazida, Rev. Bras. Farm., 2010, 91, 126-132.
23. A. R. Gennaro, J.P. Remington, Remington: a ciência e a prática da farmácia. Guanabara Koogan, Rio de Janeiro, 20ª ed., (2004).
24. R. D. C. n.31, Brasil, de 11 de agosto de 2010. Realização dos Estudos de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo. Diário Oficial da União 2010, 11 ago.
25. R. P. Moises, Tecnologia de Produção de Comprimidos. Fármacos & Medicamentos, Rev. Bras. Farm., 2006, ., 38-46.
26. L.N.C. Rodrigues, S.P. Watanabe, H.G. Ferraz, Perfil de dissolução “in vitro” de comprimidos de primaquina disponíveis para tratamento de malária no Brasil, Revista da Sociedade Brasileira de Medicina Tropical, 2008, 41, 41-45.
27. E. G. Leite, Estabilidade: importante parâmetro para avaliar a qualidade, segurança e eficácia de fármacos e medicamentos [Dissertação], Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, (2005).
28. F. J. Silva, M. H. Rodrigues, T. M. Freitas, M. V. Pinto, Controle de qualidade físico-químico de comprimidos de ibuprofeno 300 mg, Revista Faculdade Montes Belos, 2014, ., 151-162.
29. M. Azeem, H. Naureen, M. Malik, Post market in-vitro bioequivalence study on representative brands of ciprofloxacin tablets (500 mg) prescribed in typhoid disease, American Journal of Pharmacological Sciences, 2014, ., 8-1.
30. L.V. Laporta, T. F. Brum, F. R. Pons Junior, M. R. Santos, C. A. Gonçalves, Validação de método analítico para avaliação da qualidade de cápsulas de cloridrato de metformina manipuladas, Revista de Ciências Farmacêuticas Básica e Aplicada, 2013, 34, 35-244.
Autor notes