Casos clínicos
Síndrome de insensibilidad completa a los andrógenos: diagnóstico y manejo multidisciplinario
Complete androgen insensitivity syndrome: diagnosis and multidisciplinary management
 Ketzarly Gitzelim Gallardo-Durán
Beatriz Elizabeth De la Fuente-Cortez
Teresa Carolina Villarreal-Benavides
Ketzarly Gitzelim Gallardo-Durán
Beatriz Elizabeth De la Fuente-Cortez
Teresa Carolina Villarreal-Benavides
Síndrome de insensibilidad completa a los andrógenos: diagnóstico y manejo multidisciplinario
Revista Médica del Instituto Mexicano del Seguro Social, vol. 61, núm. 1, pp. 117-122, 2023
Instituto Mexicano del Seguro Social

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-SinDerivar 4.0 Internacional.
Recepción: 11 Junio 2022
Aprobación: 17 Agosto 2022
Resumen:
Introducción: el síndrome de insensibilidad completa a los andrógenos (SICA) es un desorden de la diferenciación sexual, causado por un defecto en el gen receptor de andrógenos (AR; OMIM# 313700). Se caracteriza por la resistencia de los tejidos diana a la acción de la testosterona, lo que impide el desarrollo genital masculino de manera normal. El objetivo es describir un caso familiar de SICA y destacar la importancia del manejo médico multidisciplinario y el diagnóstico temprano de este síndrome. Caso clínico: presentamos dos casos de SICA en una familia mexicana. Caso 1: paciente de 18 años con amenorrea primaria y antecedente de intervención quirúrgica a edad temprana, sin realizarle gonadectomía. Caso 2: paciente de 11 años que debido al antecedente de su hermana fue intervenida quirúrgicamente a esa edad. En ambas pacientes, se reporta ausencia de útero y ovarios, vagina hipoplásica y gónadas masculinas. El cariotipo 46,XY fue detectado con técnica de bandas GTG y CBG e hibridación in situ fluorescente con presencia del cromosoma Y en el 100% de las células analizadas. Aunque ambas se identificaban con su sexo de asignación, fueron referidas a consulta de psiquiatría de la institución. Conclusiones: se discute la importancia del manejo multidisciplinario para el diagnóstico de SICA a edades tempranas con la finalidad de tomar decisiones respecto al tratamiento y manejo de las pacientes y evitar la malignización de las gónadas masculinas.
Palabras clave: Amenorrea, Andrógenos, Manejo de Atención al Paciente, Síndrome de Resistencia Androgénica, México.
Abstract:
Background: Complete androgen insensitivity syndrome (CAIS) is a sexual differentiation disorder, caused by a defect in the androgen receptor gene (AR; OMIM# 313700). It is characterized by the resistance of target tissues to the action of testosterone, which prevents normal male genital development. The objective is to describe a family case of CAIS and highlight the importance of multidisciplinary medical management and early diagnosis of this syndrome. Clinical case: We present two cases of SICA in a Mexican family. Case 1: 18-year-old female patient with primary amenorrhea and a history of surgery at an early age, without performing gonadectomy. Case 2: 11-year-old female patient who, due to the history of her sister, underwent surgery at that age. In both patients, absence of uterus and ovaries, hypoplastic vagina and male gonads is reported. The 46,XY karyotype was detected with the GTG and CBG band technique and fluorescent in situ hybridization with the presence of the Y chromosome in 100% of the cells analyzed. Although both patients were identified with their assigned sex, they were referred to the institution's psychiatric clinic. Conclusions: The importance of multidisciplinary management for the diagnosis of SICA at an early age is discussed, in order to make decisions regarding the treatment and management of patients, avoiding malignant transformation of the male gonads.
Keywords: Amenorrhea, Androgens, Patient Care Management, Androgen Resistance Syndrome, Mexico.
Introducción
El SICA forma parte de los trastornos de la diferenciación sexual. Tiene un patrón de herencia ligado al cromosoma X y es producto de mutaciones en el gen receptor para andrógenos (AR; OMIM# 313700).1 Según la Federación Mexicana de Enfermedades Raras, se estima una prevalencia de entre 1:20,000 y 1:99,000 varones nacidos vivos.2 Las características fenotípicas pueden manifestarse en la infancia con hernia inguinal con labios protuberantes que contienen testículos o en la adolescencia con amenorrea primaria. El diagnóstico se realiza basado en la clínica de la paciente y niveles elevados de hormona luteinizante (LH) y testosterona, con poca o nula virilización y cariotipo 46,XY.3 Es importante que se haga el diagnóstico en etapas tempranas para proceder a establecer tratamientos oportunos. En estos casos, interviene un equipo multidisciplinario de profesionales de la salud.4 Los primeros en recibir a la paciente, son el área de medicina general, para posteriormente apoyarse en otras especialidades médicas como la ginecología, la genética y la psiquiatría. Asimismo, para el manejo integral intervienen otras áreas, como el laboratorio clínico, patología, servicio social y el laboratorio de citogenética. El principal riesgo de estas pacientes es desarrollar gonadoblastoma, disgerminoma o seminoma, por lo que se recomienda la búsqueda intencional de gónadas dismórficas a edades tempranas.5 Presentamos dos hermanas mexicanas de 18 y 11 años, con antecedente de hernia inguinal y amenorrea primaria como motivo de consulta. La primera de ellas fue intervenida quirúrgicamente en su infancia sin realizar gonadectomía. Discutimos la clínica de las pacientes y realzamos la importancia del manejo multidisciplinario en etapas tempranas de la enfermedad.
Caso 1
Paciente mujer, mexicana, de 18 años, II-1 (figura 1), obtenida de parto eutócico, cuyos datos pre y post natales se desconocen.
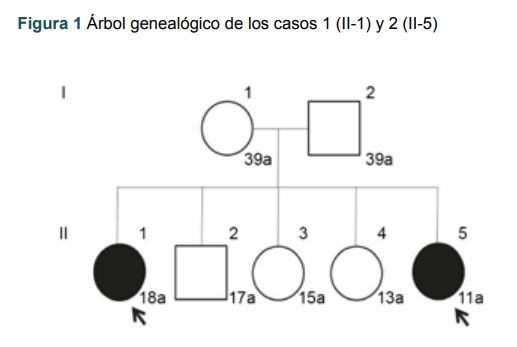
Figura 1
Árbol genealógico de los casos 1 (II-1) y 2 (II-5)
La paciente acudió a nuestra consulta de medicina general y refirió que a los cinco años fue intervenida quirúrgicamente, en otro hospital diferente al nuestro, por hernia inguinal bilateral, con gónada que no fue extirpada. A los 15 años la paciente fue manejada con terapia de reemplazo hormonal por varios meses por amenorrea primaria, sin presentar nunca menarquia; el inicio de su vida sexual fue a los 17 años con dispareunia. La paciente fue referida a ginecología, donde a la exploración física presentó tórax en dimensiones normales, con glándulas mamarias en Tanner IV, ausencia de vello axilar (figura 2) y púbico, genitales externos femeninos con hipoplasia de labios, clítoris y vagina.
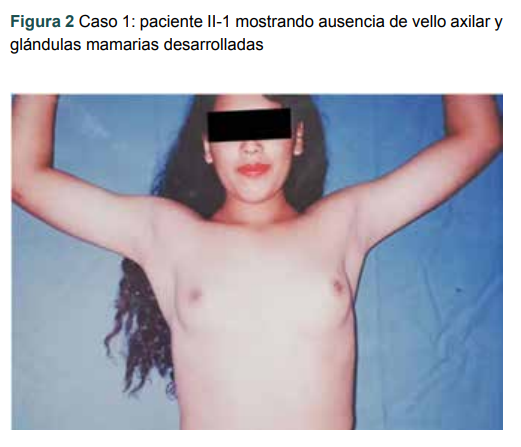
Figura 2
Caso 1: paciente II-1 mostrando ausencia de vello axilar y glándulas mamarias desarrolladas
Se le hicieron estudios clínicos de laboratorio, los cuales mostraron niveles de FSH 46.8 mUI/mL (valores de referencia: mujeres antes de la pubertad 0-5.0 UI/L, durante la pubertad: 0.3-10.0 UI/L, adultos: 1.5-12.4 UI/L; varones: antes de la pubertad 0-5.0 UI/L, durante la pubertad: 0.3-10.0 UI/L, adultos: 1.5-12.4 UI/L), LH 36.5 mUI/mL (valor de referencia: 5-25 UI/L mujeres antes de la menopausia y 1.8-8.6 UI/L varones mayores de 18 años), testosterona sérica de 250 ng/mL (valores de referencia: varones adultos 2.62-15.93 ng/mL y mujer joven: 2.2-8.0 ng/mL), estradiol 22 pg/mL (valor de referencia: mujeres antes de la menopausia: de 30-400 pg/mL, mujeres después de la menopausia: 0-30 pg/mL, varones: 10- 50 pg/mL).
El ultrasonido abdómino-pélvico evidenció ausencia de útero y rudimento de vagina, gónadas presentes en anillo inguinal superficial, de 1.3 x 3.5 x 1.1 cm en el lado derecho y 1.5 x 2.1 x 1.6 cm del lado izquierdo. A partir de estos resultados, se le hizo gonadectomía bilateral a la paciente y se enviaron ambas gónadas a estudio histopatológico, el cual reveló dos testículos con túbulos seminíferos atróficos que contenían solo células de Sertoli, asociados a hiperplasia de células de Leydig. Se pidió apoyo al departamento de citogenética, se hizo estudio cromosómico de linfocitos en sangre venosa periférica con bandas GTG (bandas G con tripsina y giemsa) (figura 3a) y bandas CBG (bandas C con bario y giemsa) (figura 3b) y se encontró fórmula cromosómica 46,XY, normal. Se realizó hibridación in situ fluorescente (FISH) con sonda específica para el cromosoma Y marcada en rojo (DYZ1, Vysis, Abbott, Illinois, USA) y sonda específica para el cromosoma X marcada en verde (DXZ1, Vysis, Abbott, Illinois, USA) utilizada como control. Se analizaron 1000 núcleos y se encontraron 100% de las células analizadas con presencia de cromosoma Y: nuc ish (DZY1, DXZ1) x 1 [1000] (figura 3c).
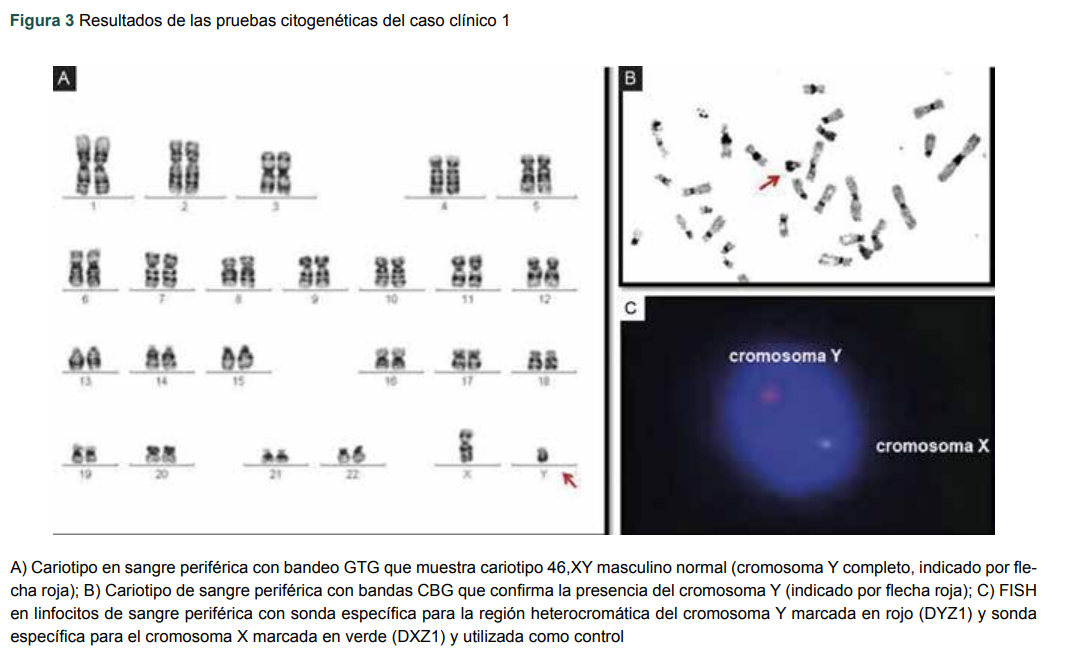
Figura 3
Resultados de las pruebas citogenéticas del caso clínico 1
a) Cariotipo en sangre periférica con bandeo GTG que muestra cariotipo 46,XY masculino normal (cromosoma Y completo, indicado por flecha roja); b) Cariotipo de sangre periférica con bandas CBG que confirma la presencia del cromosoma Y (indicado por flecha roja); c) FISH en linfocitos de sangre periférica con sonda específica para la región heterocromática del cromosoma Y marcada en rojo (DYZ1) y sonda específica para el cromosoma X marcada en verde (DXZ1) y utilizada como control
Caso 2
Paciente mujer, mexicana, de 11 años, II-5 (figura 1), hermana del caso 1, fenotípicamente femenina, quinta gesta, obtenida por cesárea. Refirió que al nacimiento se detectó masa tumoral en labio mayor izquierdo, por lo que se indicó intervención quirúrgica por sospecha de probable hernia inguinal y se encontró gónada que se reportó como testículo en el análisis histopatológico, por lo que se extirpó a su vez la gónada contralateral. A la exploración ginecológica y el ultrasonido abdómino-pélvico se encontraron genitales externos femeninos hipoplásicos, vagina pequeña y con fondo de saco con ausencia de gónadas. Ante los estudios clínicos de laboratorio, se presentaron los siguientes niveles: FSH 55 mUI/mL, LH 23.9 mUI/mL, testosterona sérica de 450 ng/mL, estradiol 18 pg/mL. El cariotipo con bandas GTG mostró complemento cromosómico 46,XY normal, con presencia de cromosoma Y completo, confirmado por bandas CBG. Se hizo FISH con sonda específica para el cromosoma Y marcada en rojo (DYZ1, Vysis, Abbott, Illinois, USA) y sonda específica para el cromosoma X marcada en verde (DXZ1, Vysis, Abbott, Illinois, USA) utilizada como control. Se analizaron 1000 núcleos y se enontraron 100% de las células analizadas con presencia de cromosoma Y: nuc ish (DZY1, DXZ1) x 1 [1000].
De acuerdo con la historia clínica, los resultados de gabinete, el laboratorio clínico y los estudios citogenéticos, el diagnóstico de ambas pacientes fue SICA. Se brindó asesoramiento por el departamento de genética tanto a las pacientes como a sus familiares. La mamá y las otras dos hermanas no pudieron ser estudiadas y no fue posible realizar el estudio molecular para detectar alguna mutación en el gen AR, lo cual es una limitante para este estudio, pero no lo fue para establecer el diagnóstico de las pacientes. Ninguna de las pacientes tenía datos de alteraciones óseas, ni sospechas de alguna alteración metabólica. La evolución postoperatoria de ambas pacientes resultó satisfactoria. En el caso 1, se inició terapia de reemplazo hormonal con estrógenos conjugados, a una dosis de 0.625 mg/día. Aunque ambas pacientes se identificaban con su sexo de asignación femenino, fueron enviadas a terapia de apoyo en el departamento de psiquiatría de la institución.
Cuestiones éticas
Ambas pacientes firmaron consentimiento informado por escrito para la publicación de sus datos, resultados y fotografías en el presente artículo.
Discusión
El diagnóstico de SICA normalmente se lleva a cabo en la adolescencia por amenorrea primaria o con menor frecuencia por hernia inguinal en la infancia.6 En nuestro primer paciente (caso 1) destaca el abordaje médico que le brindaron a los cinco años, cuando fue sometida a cirugía sin realizarle gonadectomía. Cuantos más años se conserven las gónadas, mayor será el riesgo de malignizarse,7 con un riesgo aproximado del 12%.8 Algunos autores refieren que es mejor retirarlas hasta la adolescencia que ya se hayan desarrollado los caracteres sexuales secundarios7,9 para asegurar la pubertad espontánea y la conversión de testosterona a estrógenos mediante la aromatasa.10 Cuando la gonadectomía se hace en la infancia, se induce la pubertad por medio de terapia de reemplazo hormonal para prevenir la regresión de caracteres sexuales secundarios y consecuencias debidas a la deficiencia de estrógenos, como baja densidad ósea y osteoporosis.8,10 De cualquier forma, es indicativo que la paciente esté informada de la decisión del equipo médico para manejo y seguimiento en etapas posteriores. La paciente del caso 1 solo estaba enterada de que se le había realizado una cirugía sin información de que se habían respetado las gónadas.
Llaman la atención los niveles altos de FSH y LH en esta paciente. Podemos pensar que el haber conservado sus gónadas masculinas hasta los 18 años influyó para que se elevaran los niveles de estas hormonas, ya que al no haber receptor que capte la testosterona, no hay retroalimentación negativa y siguen produciéndose las gonadotropinas.11,12
La paciente del caso 2 tuvo un diagnóstico más temprano, gracias al antecedente de su hermana, ya que para su edad (11 años) aún no se esperaría un desarrollo puberal completo.
Al analizar ambos casos, podemos definir que es de gran importancia el abordaje del médico de primer contacto, ya que su labor está basada en detectar alteraciones y referirlas a otras especialidades médicas para establecer un diagnóstico.
El manejo del SICA debe ser individualizado y lo debe hacer un equipo multidisciplinario7 enfocado en atender las necesidades funcionales, sexuales y psicosexuales de la paciente en cuanto a identidad de género, rol de género y orientación sexual, tanto para el paciente como para su familia.13 Antes de proceder al manejo es fundamental establecer los principios éticos del correcto actuar para respetar la autonomía del paciente y asegurarse de que cuente con toda la información necesaria de su padecimiento para que pueda tomar decisiones.14,15
La mayoría de las pacientes con SICA se identifican con el sexo de asignación femenino,14 pero aun así es necesario considerar la edad adecuada para poder decidir; se estima que entre los cinco y los seis años pueden tener un rol de identidad y entre los 12 y los 14 años pueden tomar decisiones autónomas. Los grupos de autoayuda que expongan experiencias personales son importantes para la aceptación y el desarrollo óptimo, así como el apoyo psicológico a familiares, ya que se ha documentado que en especial las madres experimentan sentimientos de culpa.16 Se recomienda seguimiento por el departamento de psiquiatría y acompañamiento psicológico, para evaluar una salud mental, debido al riesgo de presentar alteraciones de la psique.
Una parte importante del tratamiento de las pacientes con SICA es el remplazo hormonal, apoyados con el área de ginecología. Si la gonadectomía se realiza en la infancia, la pubertad debe ser inducida por remplazo hormonal estrogénico, por medio de etinilestradiol con una dosis inicial de 2 μg diarios desde los 11 años para después incrementar la dosis 2-4 μg por dos años hasta que se alcance la dosis de 30 μg. Una vez alcanzada la pubertad, se continúa con el curso de estrógenos ya sean orales o transdérmicos.17 Si la gonadectomía se realiza en la adolescencia, es importante vigilar por medio de estudios de imágenes que no exista malignidad de las gónadas, porque ningún marcador tumoral es detectado en la circulación.18 Las pacientes con SICA tienen estrechez del introito vaginal, lo que ocasiona dificultad a la penetración vaginal. Esto puede corregirse con dilataciones quirúrgicas. Se requiere apoyo de un ginecólogo especialista en fertilidad para explicar las opciones (como la adopción o el útero subrogado) en caso de querer lograr un embarazo.17,18
Se debe deferir al departamento de genética para brindar asesoramiento para explicar a los padres que este padecimiento es debido a una alteración genética en el cromosoma X y que existe la posibilidad de ser heredado a otras hijas por una madre portadora.18 Hay otros casos familiares reportados, similares al nuestro que realzan la importancia del diagnóstico y manejo multidisciplinario del SICA (cuadro I),19,20,21,22 ninguno de ellos en nuestro país.
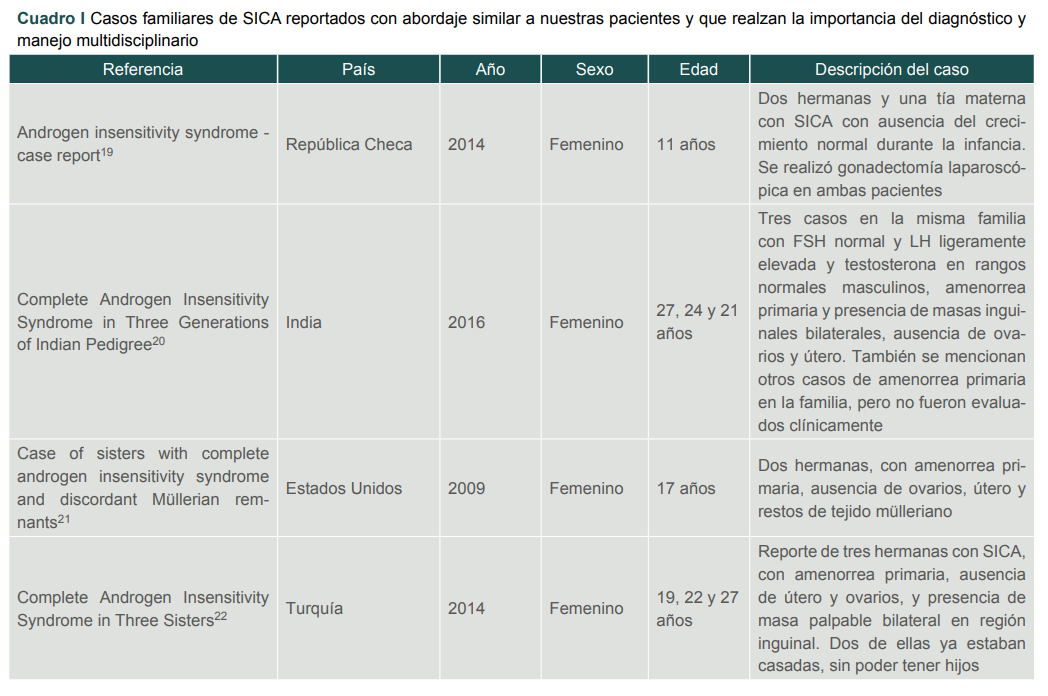
Cuadro I
Casos familiares de SICA reportados con abordaje similar a nuestras pacientes y que realzan la importancia del diagnóstico y manejo multidisciplinario
Conclusiones
Se enfatiza la importancia de estudiar adecuadamente a las pacientes con SICA y brindar información veraz y oportuna a los familiares. Es indispensable la estrecha colaboración de un equipo multidisciplinario de médicos especialistas para brindar un abordaje completo y temprano. No debe fomentarse la relación paternalista con la paciente; se debe explicar que existe discordancia del sexo de asignación con el cromosómico. Esto no define su conducta, ni su identidad sexual, ya que esta se ve más influenciada por el ambiente cultural. Lo anterior, siempre respetando la autonomía de la paciente para que, con base en la información proporcionada, pueda tomar las decisiones propias de su salud.
Referencias
1. Galani A, Kitsiou-Tzeli S, Sofokleous C, Kanavakis E, Kalpini-Mavrou A. Androgen insensitivity syndrome: clinical features and molecular defects. Hormones (Athens). 2008;7(3):217-29. doi: 10.14310/horm.2002.1201
2. Batista RL, Frade Costa EM, Rodrigues A de S, Gomes NL, Faria JA Jr, Nishi MY, et al. Androgen insensitivity syndrome: a review. Arch Endocrinol Metab. 2018;62(2):227-35. doi: 10.20945/2359-3997000000031
3. Federación Mexicana de Enfermedades Raras. Síndrome de insensibilidad completa a los andrógenos. México: Femexer.org; 30 de abril de 2014. Disponible en: http://www.femexer.org/3522/sindrome-de-insensibilidad-completa-a-los-androgenos/
4. Solana MA, Paris A. Síndrome de insensibilidad completa a los andrógenos. SAEGRE. 2013; 20(1):36-9. Disponible en: http://www.saegre.org.ar/revista/numeros/2013/n1/36-40-2013n1.pdf
5. Farias-Cortés JD, Minakata-Ochoa F, Sedano-Portillo I. Síndrome de insensibilidad completa a los andrógenos: reporte de un caso, ilustración del manejo quirúrgico. Revista Mexicana de Urología. 2014;74(2):117-22. doi: 10.1016/S2007-4085(15)30023-9
6. Mongan NP, Tadokoro-Cuccaro R, Bunch T, Hughes IA. Androgen insensitivity syndrome. Best Pract Res Clin Endocrinol Metab. 2015;29(4):569–80. doi: 10.1016/j.beem.2015.04.005
7. Li BK, Ding Q, Wan XD, Wang X. Clinical and genetic characterization of complete androgen insensitivity syndrome in a Chinese family. Genet Mol Res 2011;10(2):1022-31. doi: 10.4238/vol10-2gmr1130
8. Wisniewski AB, Migeon CJ, Meyer-Bahlburg HFL, Gearhart JP, Berkovitz GD, Brown TR, et al. Complete androgen insensitivity syndrome: long-term medical, surgical, and psychosexual outcome. J Clin Endocrinol Metab. 2000;85(8):2664-9. doi: 10.1210/jcem.85.8.6742
9. Li L, Liu WM, Liu MX, Zheng SQ, Zhang JX, Che FY, et al. A missense mutation in the androgen receptor gene causing androgen insensitivity syndrome in a Chinese family. Asian J Androl. 2017;19(2):260-1. doi: 10.4103/1008-682X.172647
10. Tadokoro-Cuccaro R, Hughes IA. Androgen insensitivity syndrome. Curr Opin Endocrinol Diabetes Obes. 2014;21(6):499-503: doi: 10.1097/MED.0000000000000107
11. Oakes MB, Eyvazzadeh AD, Quint E, Smith YR. Complete androgen insensitivity syndrome: a review. J Pediatr Adolesc Gynecol 2008;21(6):305-10. doi: 10.1016/j.jpag.2007.09.006
12. Holterhus PM, Wiebel J, Sinnecker GHG, Brüggenwirth HT, Sippell WG, Brinkmann AO, et al. Clinical and molecular spectrum of somatic mosaicism in androgen insensitivity syndrome. Pediatr Res. 1999;46(6):684-90. doi: 10.1203/00006450-199912000-00009
13. Hughes IA, Houk C, Ahmed S F, Lee P A; LWPES1/ESPE2 Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91(7):554-63. doi: 10.1136/adc.2006.098319
14. Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou, K, MacDougall J. Androgen insensitivity syndrome. Lancet. 2012;380(9851): 1419-28. doi: 10.1016/S0140-6736(12)60071-3
15. Indig G, Serrano M, Dalke KB, Nwadiogo IE, Grimstad F. Clinician Advocacy and Intersex Health: A History of Intersex Health Care and the Role of the Clinician Advocate Past, Present, and Future. Pediatric Annals. 2021;50(9):e359-65. doi: 10.3928/19382359-20210816-01
16. Bransford, CL. Reconciling Paternalism and Empowerment in Clinical Practice: An Intersubjective Perspective. Social Work. 2011;56(1):33-41. doi: 10.1093/sw/56.1.33
17. Wiesemann C, Ude-Koeller S, Sinnecker GHG, Thyen U. Ethical principles and recommendations for the medical management of differences of sex development (DSD)/intersex in children and adolescents. European Journal of Pediatrics. 2010;169(6):671-9. doi: 10.1007/s00431-009-1086-x
18. Reis E, McCarthy MW. What Hospitalists Should Know About Intersex Adults. Perspect Biol Med. 2016;59(3):391-8. doi: 10.1353/pbm.2016.0033
19. Khollová S, Hrdonková E, Pomahačová R. Androgen insensitivity syndrome - case report. Ceska Gynekol. 2014;79(1):38-42.
20. Kar B, Sivamani S, Kundavi S, Varma TR. Complete androgen insensitivity syndrome in three generations of Indian pedigree. J Obstet Gynaecol India. 2016;66(Suppl 1):358-62. doi: 10.1007/s13224-015-0736-3
21. Nichols JL, Bieber EJ, Gell JS. Case of sisters with complete androgen insensitivity syndrome and discordant Müllerian remnants. Fertil Steril. 2009;91(3):932.e15-8. doi: 10.1016/j.fertnstert.2008.09.027
22. Verim L. Complete Androgen Insensitivity Syndrome in Three Sisters. Int J Fertil Steril. 2014;7(4):353-6.
Notas de autor
Comunicación con: Catalina García Vielma Teléfono: 81 8190 4036
Información adicional
Declaración de conflicto de interés:: las autoras han completado y enviado la forma traducida al español de la declaración de conflictos potenciales de interés del Comité Internacional de Editores de Revistas Médicas, y no fue reportado alguno relacionado con este artículo.
Enlace alternativo
http://revistamedica.imss.gob.mx/editorial/index.php/revista_medica/article/view/4691/4493 (pdf)