Article
DFT and Molecular Docking Studies of Melatonin and Some Analogues Interaction with Xanthine Oxidase as a Possible Antiradical Mechanism
Estudios de DFT y acoplamiento molecular de la interacción de la melatonina y algunos análogos con la xantina oxidasa como posible mecanismo antirradical
 Minerva Martinez-Alfaro
Minerva Martinez-Alfaro
DFT and Molecular Docking Studies of Melatonin and Some Analogues Interaction with Xanthine Oxidase as a Possible Antiradical Mechanism
Journal of the Mexican Chemical Society, vol. 68, no. 1, pp. 99-112, 2024
Sociedad Química de México A.C.
Received: 28 May 2023
Accepted: 14 July 2023
Funding
Funding source: University of Guanajuato
Contract number: 268
Funding statement: Guanajuato National Laboratory (CONACYT 1237332) is acknowledged for supercomputing resources. BM acknowledges support from CONACYT for a Ph.D. scholarship (580068/296892). JR gratefully acknowledges funding from University of Guanajuato (DAIP-Convocatoria Institucional de Investigación Científica 2022, Project No. 268.)
Abstract: Melatonin (Mel) and some of its active metabolites such as N1-acetyl-5-methoxykynuramine (AMK), N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), 6-hydroxymelatonin (6OHM), and the analogues Ir and It recently designed by Galano's group, have been studied within density functional theory (DFT). The purpose is to evaluate some plausible mechanisms of action of melatonin's metabolites and analogues with the free radicals (FR): , , , and . We calculated global chemical reactivity descriptors from conceptual DFT to evaluate their antiradical properties. We used water and pentyl ethanoate as solvents to simulate the physiological conditions, modeled via the continuum solvation model based on density (SMD). We assess the following plausible mechanisms: single electrons transfer (SET), hydrogen atom transfer (HAT) and xanthine oxidase (XO) inhibition. We performed our calculations at the M06-2X/6-31+G* level of theory. The results indicate that Mel, AMK, AFMK, 6OHM, It, and Ir are good antiradicals towards the FRs: and , while It and Ir could be suitable XO inhibitors.
Keywords: Antiradical properties, Density Functional Theory, melatonin, xanthine oxidase, molecular docking.
Resumen: La melatonina (Mel) y algunos de sus metabolitos activos como N1-acetil-5-metoxiquinuramina (AMK), N1-acetil-N2-formil-5-metoxiquinuramina (AFMK), 6-hidroximelatonina (6OHM) y los análogos Ir e It, diseñados recientemente por el grupo de Galano, han sido estudiados con la teoría de funcionales de la densidad (DFT). El propósito es evaluar algunos mecanismos de acción plausibles de los metabolitos y análogos de la melatonina con los radicales libres (FR): , , , y . Calculamos los descriptores de reactividad química global a partir de DFT conceptual para evaluar sus propiedades antirradicales. Usamos agua y etanoato de pentilo como solventes para simular las condiciones fisiológicas, modeladas a través del modelo continuo de solvatación basado en la densidad (SMD). Evaluamos los siguientes mecanismos plausibles: transferencia de electrones individuales (SET), transferencia de átomos de hidrógeno (HAT) e inhibición de la xantina oxidasa (XO). Realizamos nuestros cálculos al nivel de teoría M06-2X/6-31+G*. Los resultados indican que Mel, AMK, AFMK, 6OHM, It e Ir son buenos antirradicales frente a los FRs: y , mientras que It e Ir podrían ser inhibidores adecuados de XO.
Palabras clave: Propiedades antirradicales, teoría de funcionales de la densidad, melatonina, xantina oxidasa, acoplamiento molecular.
Introduction
Reactive oxygen species (ROS) and reactive nitrogen species (RNS), namely free radicals (FRs), are continuously generated in typical biological systems by enzymes like xanthine oxidase (XO) [1, 2]. However, high levels of FRs are associated with increased oxidative stress (OS) [3]. Then, the inappropriate scavenging or inhibition of FRs has been linked with aging, inflammatory disorders, and chronic diseases [4, 5]. Nevertheless, antiradicals (ARs) molecules can interact with FRs and terminate their chain reaction by different mechanisms to prevent OS [5-7]. There is evidence that melatonin and related compounds are efficient as ARs [8-11]. The literature has extensively studied the role of melatonin (Mel) and its metabolites against OS. The different mechanisms of melatonin and its metabolites have been reported, finding that melatonin metabolites are better antioxidants than melatonin itself [11-16]. In this sense, melatonin derivatives have been sought with better antiradical properties that regenerate, are non-toxic, and do not have pro-oxidant behavior.
Melatonin and metabolites have been studied against ROS by evaluating different mechanisms of action [8, 14]. However, there is no previous research on melatonin's metabolites and analogues to their mode of interaction with FR-producing enzymes, like XO. Mel has been reported as an inhibitor of these types of enzymes by theoretical [17, 18] and experimental works [19-23]. Hence, we propose inhibiting this enzyme by melatonin's metabolites and analogues could considerably reduce oxidative stress in the cell. With this aim, we decided to study Mel, its three active metabolites N1-acetyl-5-methoxy kynuramine (AMK), N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), 6-hydroxymelatonin (6OHM), and two analogues developed by Galano's group Ir ((E)-1-methyl-3-((2-phenylhydrazine)methyl)-1H-indol-7-ol) and It ((E)-3-((2-(2-fluorophenyl)hydrazine)methyl)-1-methyl-1H-indol-7-ol)) [24,25], (Fig. 1). Moreover, some ARs may exhibit more than one mechanism; they are classified as multifunctional antiradicals [26]. Then, we studied some possible mechanisms such as XO inhibition, single electron transfer (SET), and hydrogen transfer (HAT) with FRs: , , , and . Previous works studied Mel, 6OHM, AMK, and AFMK interacting with , and through the mechanisms of action SET, HAT, and radical adduct formation [12,14,15,27,28]. Henceforth, we use these results and evaluate the interaction with and that, as far as we are aware, have not been reported in computational studies for Mel, 6OHM, AMK, AFMK, It, and Ir.
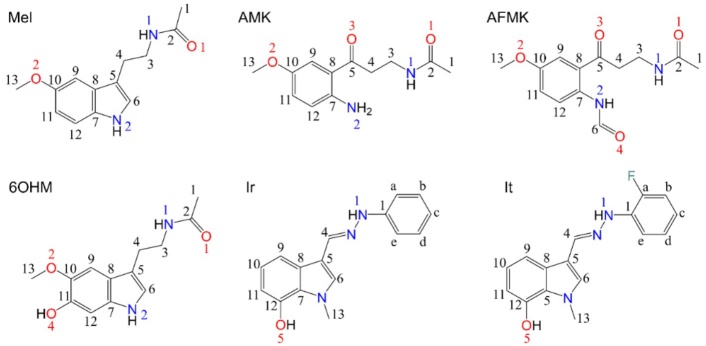
Fig. 1
Chemical structures of Mel and its analogues studied in this work and atom-numbers labels.
Consequently, we examine the antiradical properties by global chemical reactivity descriptors of Conceptual Density Functional Theory (CDFT) [29] and mechanisms (SET, HAT, and XO inhibition) of Mel, 6OHM, AMK, AFMK, It, and Ir (Fig. 1) using computer-assisted protocols that significantly reduce costs and expedite the process. The search mainly consisted of computing CDFT reactivity descriptors to assess the reactivity properties, log P, solvation free energy, and mechanisms of action of the selected species.
Computational methodology
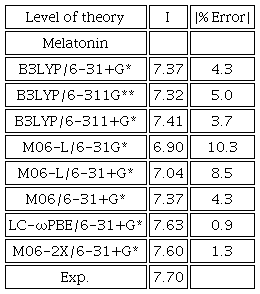
We used Gaussian 09 [30] for the molecular DFT calculations. We performed for each geometry optimization a frequency analysis to properly identify local minima in the potential energy surface (PES). The final M06-2X/6-31+G* level of theory was chosen through calibration and comparison with eight levels of theory. Since the details of the methods used are similar as in our previous work, we refer the reader to it [31]. The calculated ionization potential (I) value was used as a control and compared with the experimental I value of melatonin, 7.70 eV [32]. To simulate aqueous and lipid environments, we use the continuum solvation model based on density (SMD) [33], used to simulate solvent effects (water, ε = 78.36 and pentyl ethanoate as a model for lipidic ones, ε = 4.73).
Conformers
Initially, a conformational search is done [31,34] with the MMFF force field [35-38]. Although 10,000 conformers are inspected in the search, we select only the lowest-energy ten conformers, with a methodology detailed in our previous work [31], finally reoptimized to the level M06-2X/6-31+G* DFT.
pKadetermination
It is necessary to know the pKa value to assess the dominant acid/base species at physiological conditions (aqueous phase and average pH=7.4). Therefore, we determine the pKa values of all computed AR molecules. Since the details of the method used is similar as in our previous work, we refer the reader to it [31,39].
DFT global descriptors
Global CDFT reactivity descriptors [29] were computed to assess the reactivity of the studied molecules towards the FRs. In this work, since the details of the computational methods used are similar as in our previous work, we refer the reader to it [31]. Among them, we computed the vertical ionization energy (I) and the electron affinity (A) [40,41]. Also computed are the electronegativity (χ) [42], chemical hardness () [43,44]. Both were computed with the well-known finite difference approximations. Other calculated descriptors are the electrophilicity index (𝜔) [45], the electrodonating power index (), and the electroaccepting power index () [46].
Solvation free energy and log P
To assess the relative stability of a chemical species in solution respect to the gas phase, we computed the solvation-free Gibbs energy () [47]. Thus, we have computed the optimized Gibbs free energies in gas phase () and in the two solvents: water and pentyl ethanoate (),
We also used the log P value (octanol/water partition coefficient) to assess the hydrophobicity of compounds and their membrane permeability [48]. The log P value was computed using Spartan 18 [49] with the quantitative structure-activity relationships (QSAR) method of Ghose, Pritchet, and Crippen [50].
Single electron transfer
We used two graphical strategies to investigate the SET mechanism,
One strategy is to calculate the full-electron donor-acceptor map (FEDAM), and the second is the donator-acceptor map (DAM). FEDAM provides information about electron-donor and electron-acceptor behaviors of a given AR molecule [51]. In FEDAM one graphs I versus A to evaluate and characterize the electron-transfer process between AR and FR. The FRs evaluated were: , , , and . On the other hand, in DAM one compares different molecules and classifies them according to their electron donating-accepting capacity relative to the F and Na atomic values.
Thus, one graphs the electron acceptance () versus the electron donation index (), defined as
Hydrogen atom transfer
The HAT mechanism, is represented in general by
We studied the HAT mechanism of our selected molecules for the FRs: , , , and . Then, we computed the adiabatic Gibbs free energy for all reactions.
Also, we studied the dissociation energy of one hydrogen atom within the molecule (D0),
Molecular docking studies
To estimate the XO enzyme inhibition, we performed a flexible Docking study. We used the X-ray structure of Bos Taurus with PDB code: 1FIQ [52], (90 % homology with human XO) at 2.5 Å resolution, cocristalyzed with its competitive inhibitor salicylic acid (SAL). We employed the AutoDock Tools package version 1.5.6 and AutoDock 4.2.6 [53]. The protocol we followed was to merge nonpolar hydrogens, add Kollman charges, and use a Lamarckian Genetic Algorithm [54]. The A and B chains of the protein and small molecules were removed except for the molybdopterin cofactor (MTE) and Mo cofactor (MOS) in the C subunit of the XO protein. Since the details of the methods used are similar as in our previous work, we refer the reader to it [31]. All molecular graphics material was processed using the software Discovery Studio [55].
Results and discussion
To perform the calculations of Mel and its active metabolites, we selected the M06-2X/6-31+G* level of theory after assessing the cost/performance in several levels of theory. Since the details of the methods used are similar as in our previous work, we refer the reader to it [31]. We compare the absolute error between calculated I and experimental I, Table 1, where we see that M06-2X/6-31+G* has the second smallest absolute error. As a result, M06-2X/6-31+G* was selected for its low computational cost and similarity to the experimental data. Afterwards, we performed a conformational analysis at the M06-2X/6-31+G* level of theory.
Conformational search
The conformational analysis shows the most stable and thermodynamically more favorable conformers. Some of them have intra-molecular hydrogen bonds, Fig. 2; this may cause the highest value of and in HAT mechanism and, consequently, the lowest antioxidant potential. Then, Mel does not show any hydrogen bond in water and pentyl ethanoate, but noteworthy, the alkyl chain interacts with the benzene ring. AMK has one strong hydrogen bond between the amine and the ketone groups in both solvents. AFMK has one strong hydrogen bond and one weak hydrogen bond, where the hydrogens of the amine groups require more energy to be removed. Similarly, 6OHM has one strong hydrogen bond between the alcohol and ether groups.
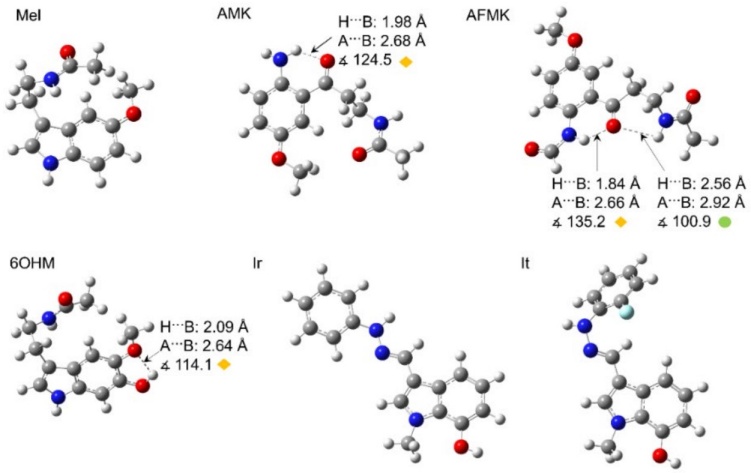
Fig. 2
Optimized geometries of Mel and its analogues in the water phase (same in pentyl ethanoate). Arrows indicate the hydrogen bonds with geometric parameter values. The colored circles and rhombuses indicate the types of hydrogen bonds formed, where yellow rhombuses show strong hydrogen bonds, and green circles show weak hydrogen bonds. Distances are reported in Å.
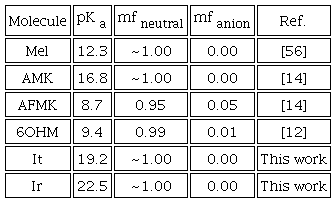
pKacalculation
The pKa values of Mel and its derivatives are displayed in Table 2, together with the corresponding molar fractions at physiological pH (7.4). Table 2 shows that the dominant species at physiological pH are neutral molecules. Consequently, all the molecules were computed in their neutral forms. Previous works report the pKa value of Mel, AMK, AFMK, and 6OHM; see Table 2. However, in this work, the pKa values of It and Ir were obtained using the direct method of the proton exchange scheme, cycle A [39] in this work. It is well known that cycle A is not very accurate for proton exchange schemes, but our results agree with experimental results, which is sufficient for the scope of this work.
Global descriptors
Our study of the reactivity of Mel and analogues has been performed by determining the global DFT reactivity descriptors, Table 3. We find that the analogues Ir and It have good antiradical activity. Furthermore, we found that Mel and analogues are more reactive in water than in pentyl ethanoate. We can see the order of molecules with the lower values of I is as follows: Ir<It<6OHM<AFMK<Mel<AMK. A lower I means a higher probability of losing an electron, thus Ir and It have higher capability for donating an electron to FRs. These results agree with the previous work of Galano [57]. In addition, we compared the molecules with lower values of A which may be ordered accordingly as: 6OHM<Mel<It<Ir<AFMK<AMK. A higher value of A means a higher capability for gaining an electron, so AFMK and AMK can accept an electron more easily from FRs.
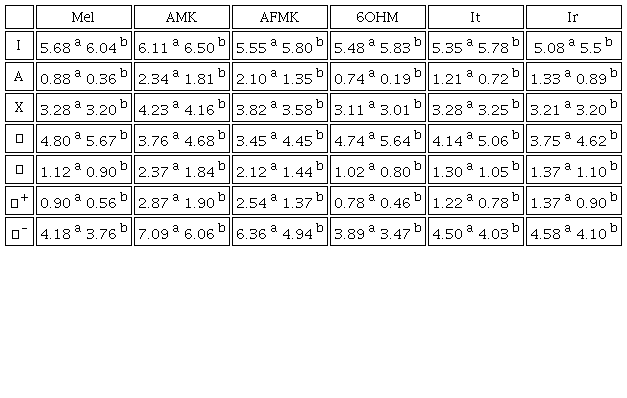
a waterb pentyl ethanoate
Furthermore, a lower value of η means low resistance to change in electron number or towards deformation of the electron cloud (higher values of 𝜂 for more stable molecules). Thus, molecules with lower values of η have the following order: AFMK<Ir<AMK<It<6OHM<Mel. Additionally, lower values mean they are more proficient in giving away electrons than capturing them. The less electronegative molecules follow the order Ir<6OHM<It<Mel<AFMK<AMK. Hence, Ir and 6OHM can donate one electron more easily.
Meanwhile, the value of 𝜔 shows that all the molecules in water except 6OHM are strong electrophiles, according to Domingo et al. [58]. These authors established an electrophilicity scale for the classification of organic molecules, defining as strong electrophiles those with 𝜔 > 1.5 eV, moderate electrophiles with 0.8 < 𝜔 < 1.5 eV, and marginal electrophiles with 𝜔 < 0.8 eV. Our molecules with the lower values of 𝜔 have the following order 6OHM<Mel<It<Ir<AFMK<AMK, where molecules with a lower value of 𝜔 are expected to be efficient for scavenging free radicals via electron transfer. Also, we see molecules with the highest value of display the following order AMK>AFMK>Ir>It>Mel>6OHM. A higher value of means a higher probability to accept charge; thus, AFMK and AMK can accept charge more easily. Molecules with lower values of display this trend: 6OHM<Mel<It<Ir<AFMK<AMK. Where, the lower value of means a higher probability to donate charge to FRs, i. e. 6OHM and Mel can donate charge more easily. This trend was also observed in the pentyl ethanoate phase.
Solvation free energy and log P
We found that melatonin and its metabolites can be classified as amphiphilic, while It and Ir are lipophilic molecules according to their log P values; see Table 4. Compounds can be classified as highly lipophilic with a value of log P > 6 (i. e. vitamin E and vitamin A); very hydrophilic with log P < -3 (i. e. vitamin C); and amphiphilic with a log P between -1 and 2 (i. e. melatonin) [59]. Although the value of indicates greater solubility in water, this may be due to the difference in solvents used to represent the lipid layer. The hydrophobicity of antiradicals helps passive transport through cell membranes. As a result, these molecules will cross the cell membrane more easily; because they are not very lipophilic, they do not tend to bioaccumulate.
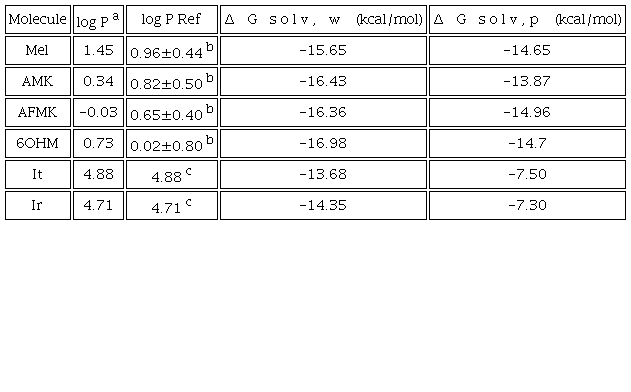
a Calculated by using Molinspiration online tool http://www.molinspiration.com.b ADC/Chemsketch log P plugin http://www.acdlabs.com in ref. [59].c Calculated by using online Molinspiration http://www.molinspiration.com in ref. [14].
In addition, melatonin and its metabolites have good bioavailability and low toxicity [57]. Ir and It have 1749 and 1795 mg/kg LD50 values, respectively. Also, the Ames mutagenicity value (M) is 0.41(-) and 0.43(-), respectively [57]. Then, It and Ir are estimated as not toxic. Moreover, according to Galano's work, these molecules are estimated to be easily synthesized. Considering that they have good bioavailability and low toxicity, different mechanisms of action have been evaluated in this work. Nevertheless, it is important to note that previous works mention that 6OHM acts against FRs as a good antiradical with primary mechanisms of action. In contrast, AMK, AFMK, and Mel act by secondary action mechanisms such as metal chelation [12,15].
Single electron transfer (SET)
We used two strategies to evaluate the SET mechanism: DAM and FEDAM maps. In the DAM map, Fig. 3, we found that all the molecules are bad acceptors and donators because they need a lot of energy to donate electron charge and have low electron affinity. As one can see, SET and SET-PT mechanisms are not favorable for all the molecules evaluated. This behavior was seen in both solvents. On the other hand, in Fig. 4, in the FEDAM, we found that AMK and AFMK can be expected to accept an electron from , especially in water. Galano et al. evaluated Mel, It, and Ir with DPPH using the FEDAM map, where they found that It and Ir are better electron acceptors than Mel [57]. In this map, the molecules down and left will transfer electrons more easily to the FR molecules located up and right, and molecules near FR can accept electrons from FRs.
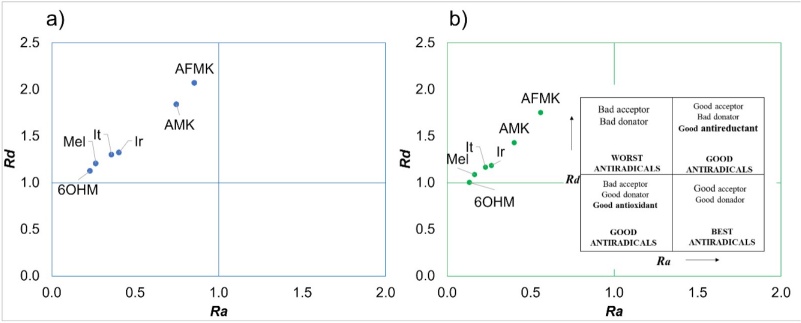
Fig. 3
DAM map of the molecules studied here where (a) molecules in water and (b) molecules in pentyl ethanoate.
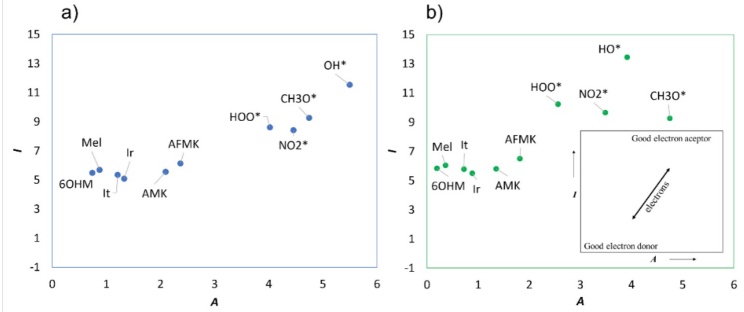
Fig. 4
FEDAM map of the molecules studied here where (a) molecules in water and (b) molecules in pentyl ethanoate.
Hydrogen atom transfer
The mechanism of hydrogen atom transfer was evaluated for all the molecules, where all of them may neutralize and . The active hydrogens for Mel and the selected metabolites are reported in previous works; in order not to repeat results, only the active hydrogen atoms already reported were considered [9,15,28,60]. On the other hand, since this was never done before, all available hydrogen atoms were studied for Ir and It.
Previous work on Mel and its metabolites [9,15,28,60] evaluated and radicals, whereas in this work, and were included. The results indicate that these molecules can neutralize FRs such as and . The best antiradical is represented by the number of reactive hydrogen atoms and the values of Δ𝐺. Negative values of Δ𝐺 indicate that the reaction is exergonic, hence thermodynamically possible. Dissociated hydrogen atoms that produce exergonic reactions are considered active hydrogens. Consequently, those molecules with several active hydrogen atoms are more reactive; therefore, they may be better antiradicals, at least by this mechanism of action.
The number of atoms studied, and the most reactive ones are displayed in Table 5. In all cases, the hydrogen atom most easily removed is the acid hydrogen shown in Fig. 2 with the lowest value, see Tables S1-S6.

() order of exergonicity - not exergonic reaction
Therefore, hydrogen bonds are important in hydrogen atom transfer since they can increase the energetic cost of removing them [61]. A strong hydrogen bond is thermodynamically more favorable; this causes a high value of and ; consequently, one has a low antiradical potential. In this case, the metabolites are those with hydrogen bonds between the acidic hydrogens, increasing their energy cost to be transferred. In Table 6, It and Ir present more exergonic reactions than Mel and its metabolites; as a result, It and Ir are assessed as good candidates as antiradicals, see more values in Tables S1-S6.
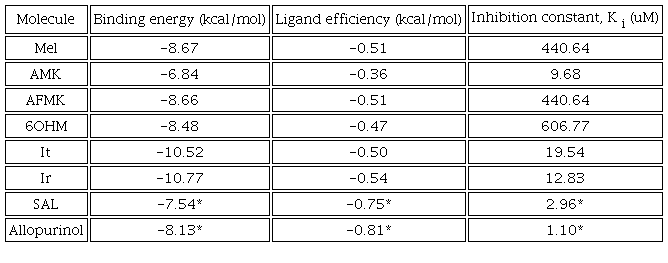
* Values taken from the ref. [31].
Inhibition of xanthine oxidase
XO is a pro-oxidative enzyme because it increases the production of ROS [2,62]. The mechanism of XO starts with the hydroxylation of hypoxanthine; then, it is converted to uric acid, releasing ROS during this catalytic process. XO contains a molybdopterin (Mo) cofactor that is responsible for oxidation. We performed a molecular docking study of XO compounds to evaluate the XO inhibition, where It and Ir could be XO inhibitors. Then, the docking study was carried out on the C subunit with the Mo cofactor [2]. For the analysis of the molecules, we included allopurinol, a molecule known to be a suitable XO inhibitor and commonly employed in the treatment of excessive uric acid and gout [63].
The docking methodology was validated by the RMSD value of the known species. The method is considered adequate if the RMSD value is smaller than 3.5 Å [64]. Therefore, docking method validation was done by redocking the natural ligand (salicylic acid (SAL)), receptor on the active site. As a result, the docked inhibitor has a 1.33 Å RMSD value. For all molecules we found that the most significant interactions are hydrogen bond, π-π stacking, and π-alkyl, see Fig. 5.
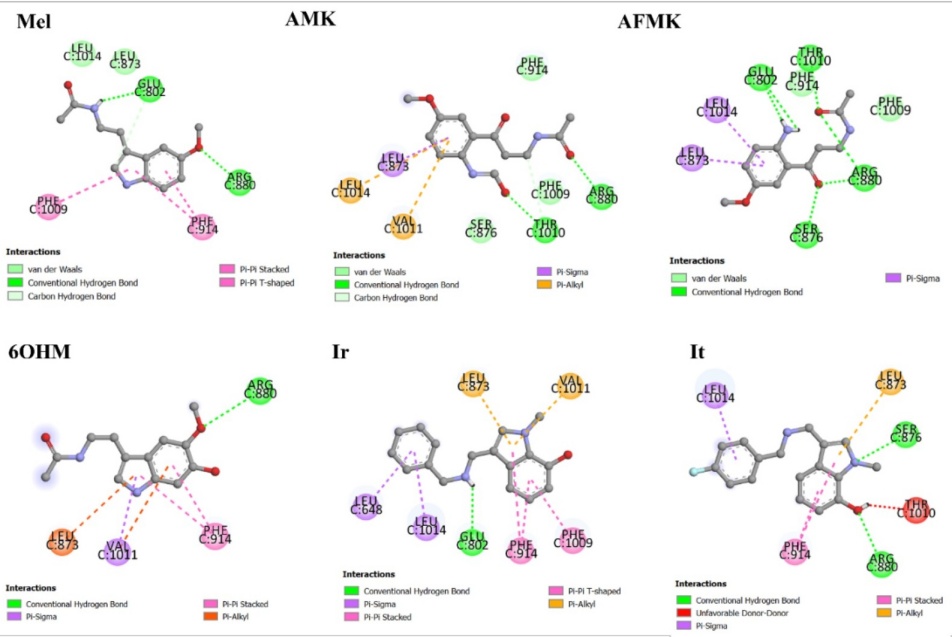
Fig. 5
2D representation of molecular interactions of Mel, metabolites, and analogues with residues of the active site of XO.
The allopurinol inhibitor and SAL were studied previously with same protocol in our previous work [31]. It is found that SAL and allopurinol exhibit strong molecular interactions with XO [31]. The binding energy and ligand efficiency reflect this, in Table 6. SAL has strong interactions with catalytic residues by a hydrogen bond with THR1010, a salt bridge with ARG880, and π-π stacking with PHE1009 and PHE914. At the same time, allopurinol has interactions with catalytic residues by π-π stacking with PHE1009 and PHE914. It is found that Mel has four significative interactions with the catalytic residues GLU802, ARG880, PHE1009, and PHE914. In the other hand, AMK has four interactions with THR1010, ARG880, LEU873, and VAL1011; AFMK has five interactions with the catalytic residues GLU802, THR1010, ARG880, LEU873, and PHE914; 6OHM has four interactions with ARG880, PHE914, VAL1011, and LEU873. Whereas Ir has six significative interactions with catalytic residues GLU802, PHE1009, LEU873, VAL1011, PHE914, and LEU648; and it has three with ARG880, LEU873, and PHE914. All observed interactions are in agreement with the results reported in other works with curcumin, tetrazoles, flavonoids, febuxostat, and analogues [65-68]. As we can see, the docking results indicated interactions with residues ARG880 and GLU802 by hydrogen bonds. The aromatic residues PHE914 and PHE1009 could interact by π-stacking, and the hydrophobic residues LEU648, LEU873, VAL1011, and LEU1014 could interact by π-sigma.
Furthermore, we found that It and Ir have the highest interaction energy and binding efficiency values and the lowest Ki value, Table 6. A high binding energy and ligand efficiency value means a stronger interaction with the target. Ligand efficiency is used to compare the activity of different molecules regardless of their sizes. On the other hand, the inhibition constant, Ki , indicates how potent an inhibitor is. Then, Ki is the concentration required to produce half-maximum inhibition. In consequence, Ir and Ir are the most promising compounds for inhibiting XO of the compounds evaluated.
Conclusions
Antiradical properties of Mel, its metabolites, and analogues proposed by Galano's group [24,25] were evaluated at the M06-2X/6-31+G(d) level of theory. This level of theory was selected considering the best compromise time/performance, after calibration of several levels of theory and using the experimental value of the vertical ionization potential of Mel as a reference. Furthermore, we evaluated some possible mechanisms of action such as SET, HAT, and XO inhibition.
The SET possible mechanisms of Mel, metabolites, and analogues were evaluated, and we computed the CDFT chemical descriptors. 6OHM, Mel, It, and Ir are the best donating electronic charges to FRs. Subsequently, the SET mechanism was evaluated using DAM and FEDAM tools, where the molecules showed resistance to transfer or accept electronic charge. On the other hand, we computed the HAT mechanism finding that the molecules show favorable results to inhibit FRs: , , , and . In this work, we suggest that Mel, metabolites, and their analogues Ir and It, may neutralize the FRs and . Finally, It and Ir appear to be suitable inhibitors of XO since they have relatively good value of ligand efficiency, compared to that of the inhibitors: allopurinol and SAL, and a low value of the Ki . Some of the conclusions that are drawn from our extensive docking studies are in good agreement with what would intuitively be expected. Nevertheless, we believe that the amount of quantitative data and results presented, fully support the conclusions.
Noteworthy, other mechanisms of action, such as electron-proton sequence transfer (SET-PT), can be anticipated with the help of the global reactivity descriptors. The high I values, indicate that the studied molecules are unlikely to exhibit the SET-PT mechanism in physiological and lipophilic media. The same applies to the sequential proton-losing hydrogen atom transfer (SPLHAT) mechanism. Still, other mechanisms of primary, secondary, and tertiary action need to be evaluated to complete our study of these molecules as antiradicals and their possible active metabolites. Therefore, due to the very favorable values obtained of low toxicity, good solubility, and accessible synthesis, Ir and It may be recommended as good synthetic antiradicals against oxidative stress.
Acknowledgements
Guanajuato National Laboratory (CONACYT 1237332) is acknowledged for supercomputing resources. BM acknowledges support from CONACYT for a Ph.D. scholarship (580068/296892). JR gratefully acknowledges funding from University of Guanajuato (DAIP-Convocatoria Institucional de Investigación Científica 2022, Project No. 268.)
References
Dharmaraja, A. T. J. Med. Chem. 2017, 60, 3221-3240. DOI: https://doi.org/10.1021/acs.jmedchem.6b01243.
Kostić, D. A.; Dimitrijević, D. S.; Stojanović, G. S.; Palić, I. R.; Đorđević, A. S.; Ickovski, J. D. J. Chem. 2015, 2015, 294858. DOI: https://doi.org/10.1155/2015/294858.
Giorgi, C.; Marchi, S.; Simoes, I. C. M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Chapter Six. in: Mitochondria and Longevity; López-Otín, C., Galluzzi, L. B. T.-I. R. of C. and M. B., Eds.; Academic Press, 2018; 340, 209-344 DOI: https://doi.org/https://doi.org/10.1016/bs.ircmb.2018.05.006.
Phaniendra, A.; Jestadi, D. B.; Periyasamy, L. Indian J. Clin. Biochem. 2015, 30, 11-26. DOI: https://doi.org/10.1007/s12291-014-0446-0.
Alkadi, H. Infect. Disord. Drug. Targets. 2020, 20, 16-26. DOI: https://doi.org/10.2174/1871526518666180628124323.
Oroian, M.; Escriche, I. Int. Food Res. J. 2015, 74, 10-36. DOI: https://doi.org/10.1016/j.foodres.2015.04.018.
Jamshidi-kia, F.; Wibowo, J. P.; Elachouri, M.; Masumi, R.; Salehifard-Jouneghani, A.; Abolhasanzadeh, Z.; Lorigooini, Z. J. Herbmed. Pharmacol. 2020, 9, 191-199. DOI: https://doi.org/10.34172/jhp.2020.25.
Galano, A.; Tan, D. X.; Reiter, R. J. J. Pineal Res. 2011, 51, 1-16. DOI: https://doi.org/10.1111/j.1600-079X.2011.00916.x.
Galano, A. RSC Adv. 2016, 6, 22951-22963. DOI: https://doi.org/10.1039/c6ra00549g.
Poeggeler, B.; Saarela, S.; Reiter, R. J.; Tan, D. X.; Chen, L. D.; Manchester, L. C.; Barlow-Walden, L. R. Ann. N. Y. Acad. Sci. 1994, 738, 419-420. DOI: https://doi.org/10.1111/j.1749-6632.1994.tb21831.x.
Galano, A.; Reiter, R. J. J. Pineal Res. 2018, 65, e12514. DOI: https://doi.org/https://doi.org/10.1111/jpi.12514.
Álvarez-Diduk, R.; Galano, A.; Tan, D. X.; Reiter, R. J. Theor. Chem. Acc. 2016, 135, 38. DOI: https://doi.org/10.1007/s00214-015-1785-5.
Ramis, M. R.; Esteban, S.; Miralles, A.; Tan, D.-X.; Reiter, R. J. Curr. Med. Chem, 2015, 22, 2690-2711. DOI: https://doi.org/10.2174/0929867322666150619104143.
Galano, A.; Tan, D. X.; Reiter, R. J. J. Pineal Res. 2013, 54, 245-257. DOI: https://doi.org/10.1111/jpi.12010.
Álvarez-Diduk, R.; Galano, A.; Tan, D. X.; Reiter, R. J. J. Phys. Chem. B. 2015, 119, 8535-8543. DOI: https://doi.org/10.1021/acs.jpcb.5b04920.
Hardeland, R. Endocrine. 2005, 27, 119-130. DOI: https://doi.org/10.1385/endo:27:2:119.
Costa, J. D.; Ramos, R. D.; Costa, K. D.; Brasil, D. D.; Silva, C. H.; Ferreira, E. F.; Borges, R. D.; Campos, J. M.; Macêdo, W. J.; Santos, C. B. Mol. 2018. DOI: https://doi.org/10.3390/molecules23112801.
Guerra-Vargas, M. A.; Rosales-Hernández, M. C.; Martínez-Fonseca, N.; Padilla-Martínez, I.; Fonseca-Sabater, Y.; Martínez-Ramos, F. Med. Chem. Res. 2018, 27, 1186-1197. DOI: https://doi.org/10.1007/s00044-018-2139-3.
Kaçmaz, A.; User, E. Y.; Şehirli, A. Ö.; Tilki, M.; Ozkan, S.; Şener, G. Surg. Today. 2005, 35, 744-750. DOI: https://doi.org/10.1007/s00595-005-3027-2.
Okutan, H.; Savas, C.; Delibas, N. Interact. Cardiovasc. Thorac. Surg. 2004, 3, 519-522. DOI: https://doi.org/10.1016/j.icvts.2004.05.005.
Juan, C. A.; Pérez de la Lastra, J. M.; Plou, F. J.; Pérez-Lebeña, E. Int. J. Mol. Sci. 2021. DOI: https://doi.org/10.3390/ijms22094642.
Teixeira, A.; Morfim, M. P.; de Cordova, C. A. S.; Charão, C. C. T.; de Lima, V. R.; Creczynski-Pasa, T. B. J. Pineal Res. 2003, 35, 262-268. DOI: https://doi.org/https://doi.org/10.1034/j.1600-079X.2003.00085.x.
Zhou, J.; Zhang, S.; Zhao, X.; Wei, T. J. Pineal Res. 2008, 45, 157-165. DOI: https://doi.org/https://doi.org/10.1111/j.1600-079X.2008.00570.x.
Castañeda-Arriaga, R.; Pérez-González, A.; Reina, M.; Galano, A. Theor. Chem. Acc. 2020, 139, 1-12. DOI: https://doi.org/10.1007/s00214-020-02641-9.
Reina, M.; Castañeda-Arriaga, R.; Perez-Gonzalez, A.; Guzman-Lopez, E. G.; Tan, D.-X.; Reiter, R. J.; Galano, A. Melatonin Res. 2018, 1, 27-58. DOI: https://doi.org/10.32794/mr11250003.
Galano, A.; Raúl Alvarez-Idaboy, J. Int. J. Quantum. Chem. 2019, 119, e25665. DOI: https://doi.org/10.1002/qua.25665.
Reina, M.; Martínez, A. Comput. Theor. Chem. 2018, 1123, 111-118. DOI: https://doi.org/https://doi.org/10.1016/j.comptc.2017.11.017.
Galano, A. Phys. Chem. Chem. Phys. 2011, 13, 7178-7188. DOI: https://doi.org/10.1039/c0cp02801k.
Geerlings, P.; De Proft, F.; Langenaeker, W. Chem. Rev. 2003, 103, 1793-1874. DOI: https://doi.org/10.1021/cr990029p.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; et al. Gaussian 09. Revision C.01. Gaussian 09. Revision C.01, Gaussian, Inc, Wallingford CT. Gaussian, Inc.: Wallingford CT 2010.
Manzanilla, B.; Robles, J. J. Mol. Model. 2022, 28, 68. DOI: https://doi.org/10.1007/s00894-022-05056-4.
Cannington, P. H.; Ham, N. S. J. Electron. Spectrosc. Relat. Phenom. 1983, 32, 139-151. DOI: https://doi.org/10.1016/0368-2048(83)85092-0.
Marenich, A. V; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B. 2009, 113, 6378-6396. DOI: https://doi.org/10.1021/jp810292n.
Spartan, W. I. Spartan 08. Irvine, CA. 2008.
Halgren, T. A. J. Comput. Chem. 1996, 17, 490-519. DOI: https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P.
Halgren, T. A. J. Comput. Chem. 1996, 17, 520-552. DOI: https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<520::AID-JCC2>3.0.CO;2-W.
Halgren, T. A. J. Comput. Chem. 1996, 17, 553-586. DOI: https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<553::AID-JCC3>3.0.CO;2-T.
Halgren, T. A.; Nachbar, R. B. J. Comput. Chem. 1996, 17, 587-615. DOI: https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<587::AID-JCC4>3.0.CO;2-Q.
Ho, J.; Coote, M. L. Theor. Chem. Acc. 2009, 125, 3-21. DOI: https://doi.org/10.1007/s00214-009-0667-0.
Janak, J. F. Phys. Rev. B. 1978, 18, 7165-7168. DOI: https://doi.org/10.1103/PhysRevB.18.7165.
Casida, M. E. Phys. Rev. B. 1999, 59, 4694-4698. DOI: https://doi.org/10.1103/PhysRevB.59.4694.
Saha, B.; Bhattacharyya, P. K. RSC Adv. 2016, 6, 79768-79780. DOI: https://doi.org/10.1039/C6RA15016K.
Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512-7516. DOI: https://doi.org/10.1021/ja00364a005.
Yang, W.; Parr, R. G. PNAS. 1985, 82, 6723-6726 DOI: https://doi.org/10.1073/pnas.82.20.6723.
Parr, R. G.; Yang, W. J. Am. Chem. Soc. 1984, 106, 4049-4050. DOI: https://doi.org/10.1021/ja00326a036.
Gázquez, J. L.; Cedillo, A.; Vela, A. J. Phys. Chem. A. 2007, 111, 1966-1970. DOI: https://doi.org/10.1021/jp065459f.
Duarte Ramos Matos, G.; Kyu, D. Y.; Loeffler, H. H.; Chodera, J. D.; Shirts, M. R.; Mobley, D. L. J. Chem. Eng. Data. 2017, 62,1559-1569. DOI: https://doi.org/10.1021/acs.jced.7b00104.
Leo, A. J. Chem. Rev. 1993, 93, 1281-1306. DOI: https://doi.org/10.1021/cr00020a001.
Spartan, W. I. Spartan 18. Irvine, CA. 2018.
Ghose, A. K.; Pritchett, A.; Crippen, G. M. J. Comput. Chem. 1988, 9, 80-90. DOI: https://doi.org/10.1002/jcc.540090111.
Martínez, A.; Vargas, R.; Galano, A. J. Phys. Chem. B. 2009, 113, 12113-12120. DOI: https://doi.org/10.1021/jp903958h.
Enroth, C.; Eger, B. T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E. F. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10723-10728. DOI: https://doi.org/10.1073/pnas.97.20.10723.
Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D. S.; Olson, A. J. J. Comput. Chem. 2009, 30, 2785-2791. DOI: https://doi.org/10.1002/jcc.21256.
Morris, G. M.; Goodsell, D. S.; Halliday, R. S.; Huey, R.; Hart, W. E.; Belew, R. K.; Olson, A. J. J. Comput. Chem. 1998, 19, 1639-1662. DOI: https://doi.org/10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B.
BIOVIA. Discovery Studio Visualizer. Dassault Systèmes: San Diego 2020.
Mahal, H. S.; Sharma, H. S.; Mukherjee, T. Free Radic. Biol. Med. 1999, 26, 557-565. DOI: https://doi.org/10.1016/s0891-5849(98)00226-3.
Galano, A. A. Theor. Chem. Acc. 2016, 135, 157. DOI: https://doi.org/10.1007/s00214-016-1917-6.
Domingo, L. R.; Aurell, M. J.; Pérez, P.; Contreras, R. Tetrahedron. 2002, 58, 4417-4423. DOI: https://doi.org/https://doi.org/10.1016/S0040-4020(02)00410-6.
Johns, J. R.; Platts, J. A. Org. Biomol. Chem. 2014, 12, 7820-7827. DOI: https://doi.org/10.1039/c4ob01396d.
Galano, A.; Tan, D. X.; Reiter, R. J. RSC Adv. 2014, 4, 5220-5227. DOI: https://doi.org/10.1039/c3ra44604b.
Romero, Y.; Martínez, A. J. Mol. Model. 2015, 21, 220. DOI: https://doi.org/10.1007/s00894-015-2773-3.
Chung, H. Y.; Baek, B. S.; Song, S. H.; Kim, M. S.; Huh, J. I.; Shim, K. H.; Kim, K. W.; Lee, K. H. Age (Omaha). 1997, 20, 127-140. DOI: https://doi.org/10.1007/s11357-997-0012-2.
Lin, H.-C.; Tsai, S.-H.; Chen, C.-S.; Chang, Y.-C.; Lee, C.-M.; Lai, Z.-Y.; Lin, C.-M. Biochem. Pharmacol. 2008, 75, 1416-1425. DOI: https://doi.org/10.1016/j.bcp.2007.11.023.
Kontoyianni, M.; McClellan, L. M.; Sokol, G. S. J. Med. Chem. 2004, 47, 558-565. DOI: https://doi.org/10.1021/jm0302997.
Chen, Y.; Gao, Y.; Wu, F.; Luo, X.; Ju, X.; Liu, G. New J. Chem. 2020, 44, 19276-19287. DOI: https://doi.org/10.1039/D0NJ03221B.
Shen, L.; Ji, H.-F. Bioorg. Med. Chem. Lett. 2009, 19, 5990-5993. DOI: https://doi.org/10.1016/j.bmcl.2009.09.076.
Fatima, I.; Zafar, H.; Khan, K. M.; Saad, S. M.; Javaid, S.; Perveen, S.; Choudhary, M. I. Bioorg. Chem. 2018, 79, 201-211. DOI: https://doi.org/10.1016/j.bioorg.2018.04.021.
Santi, M. D.; Paulino Zunini, M.; Vera, B.; Bouzidi, C.; Dumontet, V.; Abin-Carriquiry, A.; Grougnet, R.; Ortega, M. G. Eur. J. Org. Chem. 2018, 143, 577-582. DOI: https://doi.org/10.1016/j.ejmech.2017.11.071.
Notes
Author notes
*Corresponding author: roblesj@ugto.mx
Conflict of interest declaration