Artículos
Rabdomiólisis: bases moleculares y presentaciones clínicas
Rhabdomyolysis: molecular bases and clinical presentation
Rabdomiólisis: bases moleculares y presentaciones clínicas
Archivos Venezolanos de Farmacología y Terapéutica, vol. 38, núm. 2, pp. 145-154, 2018
Sociedad Venezolana de Farmacología Clínica y Terapéutica
Resumen: Se define rabdomiólisis como la muerte de las fibras musculares estriadas, un fenómeno que puede obedecer a múltiples causas de diversa índole, incluyendo agotamiento energético del miocito, lesión física del mismo, miotoxicidad directa por fármacos, toxinas, procesos infecciosos e inflamatorios, drogas ilícitas, trastornos hidroelectrolíticos y endocrinos. A pesar de la heterogeneidad de su etiología, los procesos moleculares subyacentes a la rabdomiólisis tienden a converger en la disregulación del metabolismo intracelular del calcio –el cual es esencial para la contracción muscular–, que conlleva a la activación de lipasas, proteasas, disfunción mitocondrial y estrés oxidativo, finalizando en la muerte del miocito. La rabdomiólisis se acompaña de un síndrome clínico amplia- mente variable, caracterizado por desequilibrios electrolíticos, elevación de creatinkinasa sérica, y especialmente insuficiencia renal aguda debido al efecto nefrotóxico ejercido por la mioglobina liberada de los miocitos lesionados. Esta revisión describe los mecanismos fisiopatológicos observa- dos durante la rabdomiólisis y su impacto en la función renal.
Palabras clave: Rabdomiólisis, Muerte Celular, Fibra Muscular, Músculo Estriado, Mioglobina, Insuficiencia Renal Aguda.
Abstract: Rhabdomyolisis is defined as the death of striated muscle fibers, a phenomenon that may occur in response to various causes, including myocyte energy depletion, physical cell injury, direct myotoxicity by drugs, toxins and infectious and inflammatory processes; drugs of abuse, hydroelectrolytic and endocrine disorders. Despite the widely heterogeneous etiologies, the molecular mechanisms underlying rhabdomy- olysis tend to converge in dysregulation of intracellular calcium metabolism –which is essential for muscle contraction–, leading to activation of lipases and proteases, mitochondrial dysfunction and oxidative stress, finalizing in myocyte death. Rhabdomyolysis is accompanied by a broadly variable clinical syndrome characterized by electrolytic disorders, elevated serum creatine kinase, and especially acute kidney injury due to the nephrotoxic effect exerted by myoglobin released from destroyed myocytes. This review describes the patho- physiologic pathways observed during rhabdomyolisis and its impact on renal function.
Keywords: Rhabdomyolysis, Cell Death, Muscle Fiber, Striated Muscle, Myoglobin, Acute Kidney Injury.
Introducción
La etimología del término “rabdomiólisis” deriva del griego rabdo- “estriado”, myo- “músculo”, y –lisis, “descomposición”, lo cual describe el proceso de muerte celular de las fibras musculares estriadas esqueléticas, el cual se considera altamente complejo y que obedece a múltiples causas y suele acompañarse de un síndrome clínico característico generado por la presencia de diversos componentes intracelulares lesivos en la circulación sanguínea1. El síndrome ha sido observado desde tiempos bíblicos2, en el Antiguo Testamento se hace referencia a una plaga que afecto a los Israelitas durante su éxodo desde Egipto posterior al consumo abundante de codornices (Libro Números 11:31-35), donde el consumo de estas aves conlleva a miólisis debido a la intoxicación con la hierba conium maculatum, consumida por las codornices durante su migración en primavera3.
Descripciones más detalladas se encuentran en la literatura médica del siglo pasado, reportadas por Bywaters et al.,4 durante la II Guerra Mundial, describiendo un grupo de cuatro pacientes que quedaron sepultados bajo escombros tras el bombardeo Nazi a Londres en 1940; estos pacientes seguían una evolución similar, en la cual habían permanecido enterrados durante varias horas, ejerciendo presión sobre una extremidad. Al principio solo exhibían inflamación, parestesia e hipoalgesia del área afectada y lesiones cutáneas tipo ronchas o urticaria; posteriormente se observó necrosis progresiva de grandes masas musculares comprometidas en el aplastamiento. Eventualmente estos pacientes fallecieron debido a falla renal aguda, causa comprobada durante la evaluación post-mortem, donde se evidenció lesión renal de tipo degenerativa en el túbulo contorneado proximal, mientras que en la porción más distal de la nefrona se apreciaba un pigmento marrón -mioglobina- en preparaciones sin colorear similar a la de los corpúsculos sanguíneos.
La rabdomiólisis (RM) ocurre aproximadamente en un 85% de los pacientes con lesiones traumáticas5, aunque existen otras causas menos comunes como lo son las deficiencias enzimáticas, anomalías electrolíticas, enfermedades infecciosas, medicamentos, toxinas y endocrinopatías6. En cualquiera de los casos, la ruptura de la membrana de las células musculares y la eventual elevación plasmática de los componentes intra-miociticos, conlleva a una presentación clínica que puede variar desde una forma asintomática con elevación de la creatin kinasa (CK) hasta cuadros más severos con alteraciones electrolíticas, hipovolemia, acidosis metabólica, trastornos de la coagulación y falla renal aguda asociada a depósito de mioglobina7.
Esta revisión pretende resumir los aspectos estructurales y fisiológicos básicos del miocito estriado esquelético, y describir los mecanismos moleculares implicados en el fenómeno celular de la RM.
El calcio como regulador clave de la fisiología de la fibra muscular
El músculo esquelético representa aproximadamente el 40% de la masa corporal total y desempeña gran variedad de funciones, entre las cuales destacan su rol indispensable en el metabolismo –constituyendo aproximadamente 30% del gasto metabólico basal en adultos8– y su papel como generador de energía motriz en el sistema osteomuscular, permitiendo la locomoción9. En este aspecto, el calcio iónico (Ca2+) juega un rol clave, no sólo como acoplador del proceso fisiológico de la excitación-contracción muscular, sino como mediador fisiopatológico en la muerte de la fibra muscular10.
Cada fibra muscular (o miocito) contiene en su citoplasma (o sarcoplasma) miofibrillas, conjuntos de proteínas altamente organizadas en filamentos gruesos y delgados, constituidos por miosina y actina, respectivamente11. El deslizamiento de los filamentos de actina sobre los de miosina origina el acor- tamiento de la fibra observado durante la contracción11. Este deslizamiento es dirigido por el “golpe de poder” que sucede a través de la capacidad ATPasa de las cabezas de la miosina tras su unión a la actina12. Sin embargo, en estado de reposo los sitios de unión a miosina están cubiertos por el complejo tropomiosina-troponina12,13. En este escenario, el Ca2+ es esencial ya que su unión a la subunidad C de la troponina es la que origina modificaciones en su estructura que permiten la revelación de los sitios de unión a miosina en la superficie de la actina13.
Como eslabón clave en la contracción muscular, el Ca2+ está sujeto a varios sistemas de estricta regulación. En condiciones de reposo, el sarcoplasma posee una concentración de Ca2+ insuficiente para propiciar la contracción, la cual se eleva exponencialmente ante el desencadenamiento de la contracción14. Simultáneamente, la bomba antiportadora 2Na+/ Ca2+ ATPasa mantiene este estado exportando un ion Ca2+ al espacio extracelular (EEC) a cambio de dos iones Na+ que ingresan al espacio intracelular (EIC)15. En contraste, para la contracción se requiere el flujo masivo de Ca2+ hacia el sarcoplasma, proveniente del sistema sarcotubular (SS) – constituido por el retículo sarcoplásmico (RS) y los túbulos T (TT)– que es posible gracias a su disposición espacial y funcional16. Los TT inician como una continuación de la membrana celular del miocito o sarcolema, extendiéndose de manera transversal al eje de las fibras musculares que envuelven, asemejando una red perforada por estas células. Asimismo, cada TT es adyacente a los túbulos longitudinales que conforman al RS que también rodean a los miocitos, formando una cortina irregular17. Paralelamente a ambos lados de los TT, los túbulos longitudinales del RS convergen y conforman estructuras igualmente transversas, denominadas cisternas terminales, que poseen grandes depósitos de Ca2+ asociado a calsecuestrina, una proteína de unión con baja afinidad, permitiendo su rápida disociación y transporte18.
Estos depósitos de Ca2+ son liberados al sarcoplasma ante la presencia de un potencial de acción, representando el fundamento del fenómeno de acoplamiento excitación-contracción10. En este sentido, el potencial de acción que aparece en el sarcoplasma posterior a estimulación por una moto- neurona –causado por el influjo de Na+– puede propagarse rápidamente a través de los TT gracias a su situación como extensiones del sarcolema. De esta manera, los TT pueden distribuir el potencial de acción no sólo a todos los miocitos envueltos por el SS, sino también a través del RS hasta las cisternas terminales, donde generan la liberación del calcio hacia el sarcoplasma16. Este flujo sucede mediante el receptor de dihidropiridina, un canal de Ca2+ voltaje-dependiente muscular y fundamentalmente dictan las propiedades motrices de los mismos24. Alteraciones estructurales o funcionales en alguno de los componentes celulares y energéticos que aseguran la apropiada circulación de Ca2+ en el complejo miocito-SS pueden predisponer o precipitar el inicio de la RM25.
En este escenario la mioglobina juega un papel fundamental, como almacenadora y donadora esencial de oxígeno23. Esta es una hemoproteína citoplasmática de alto peso molecular26 que permite el paso de este ión ante la despolarización del 17.200 daltons constituida por una cadena polipeptídica SS19. El receptor de rianodina, un canal de Ca2+ no voltaje- dependiente, parece depender de un ligando, que podría ser el mismo calcio iónico en el sarcoplasma20; ver (Figura 1).
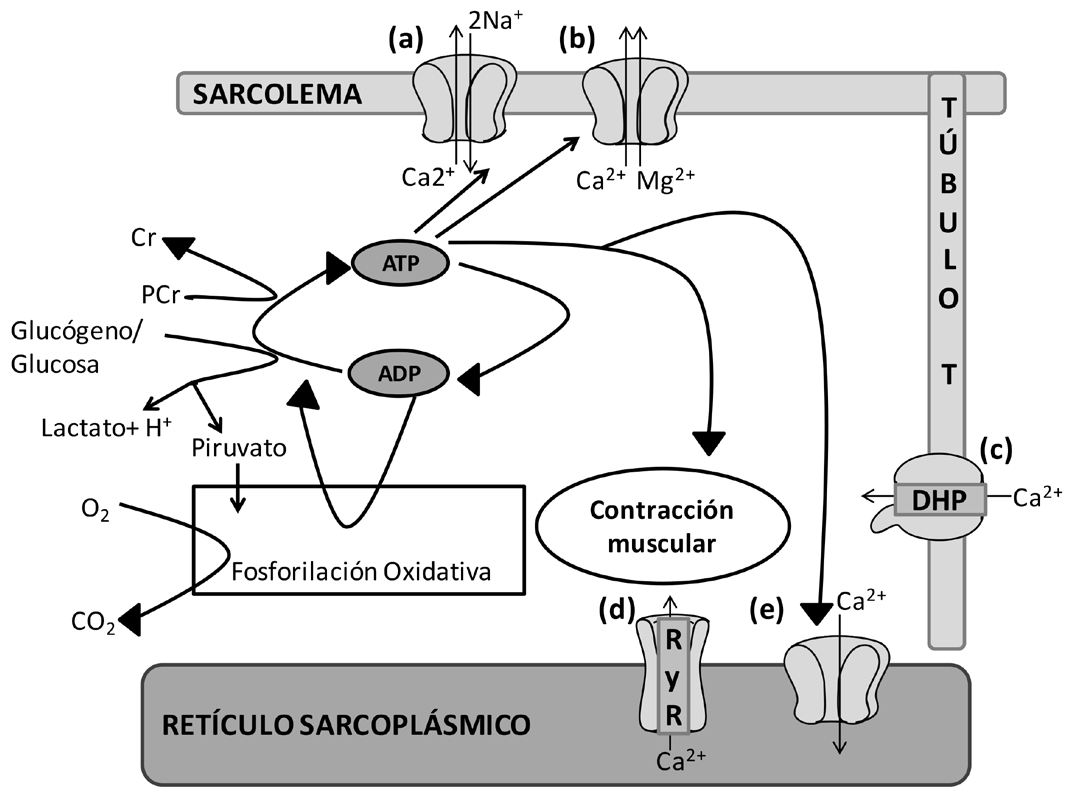
Figura 1. Representación esquemática del transporte del calcio dentro del músculo y relación entre la contracción muscular, el transporte de iones y requerimientos energéticos.
Las concentraciones basales de calcio son mantenidas por la 2Na+/Ca2+ ATPasa. Durante la contracción hay flujo masivo de este ion a través de los receptores de dihidropiridina y de rianodina. La contracción finaliza con el retorno de los niveles de Ca2+ a niveles basales, por la Ca2+ /Mg2+ ATPasa y la Ca2+ ATPasa. El ATP es la molécula esencial durante la contracción, sus demandas son cubiertas por los sistemas donadores de fosfato.
(a) 2Na+/Ca2+ ATPasa, (b) Ca2+ /Mg2+ ATPasa (c) Receptor de dihidropiridina, (d) Receptor de rianodina, (e) Ca2+ ATPasa Cr: creatina, PCr: fosfocreatina.
única de 154 aminoácidos, organizada en 8 α-hélices que encierran al grupo prostético hemo, que se localiza entre dos residuos de histidina, His64 y His93. Es este residuo hemo el que capta el oxígeno, en complejos anillo de porfirina: iones de hierro27. La mioglobina es expresada sólo encardiomiocitos y fibras musculares esqueléticas oxidativas28. La mioglobina, recibe su nombre por las similitudes estructurales y funcionales con la hemoglobina, e incluso parecen descender de un gen ancestral común29. Ambas captan oxígeno de manera reversible y facilitan su transporte hacia la mitocondria ante un incremento en la demanda metabólica o ante estados hipóxicos; no obstante, la mioglobina se une al oxígeno mucho más ávidamente, y la cinética de su disociación es escasamente modificable por factores ambientales, a diferencia de la curva de disociación oxígeno-hemoglobina30
Etiología de la rabdomiólisis: múltiples rutas a un mismo destino
La disrupción del tráfico de Ca2+ es el punto de convergencia de la mayoría de las causas de RM, desencadenando la activación de cascadas enzimáticas pro-muerte celular31. Estas etiologías pueden clasificarse artificialmente en RM por trauma físico y RM por agotamiento energético, según los rasgos fisiopatológicos que predominan en cada situación (Tabla 1). Adicionalmente, una extensa tercera categoría engloba un grupo muy heterogéneo de causas que, si bien suelen depender de la depleción energética en proporciones variables, tienden a ser multifactoriales e incluir mecanismos miotóxicos directos e independientes.
| Tabla 1. Etiología de la rabdomiólisis. Por Lesión Física Lesión por Aplastamiento Inmovilización Prolongada Quemaduras Lesión por Corriente Eléctrica Desastres Naturales Accidentes Antropogénicos Abuso y Tortura Por Agotamiento Energético Actividad Física/Ejercicio Intenso Hipoxia/Isquemia Golpe de Calor Hipertermia Maligna Síndrome Neuroléptico Maligno Agitación Psicomotriz Estatus Epiléptico Estatus Asmático Miopatías Congénitas Desacoplantes Respiratorios Miotoxicidad Multifactorial Alcohol Etílico Cocaína Anfetaminas Fenilciclidinas Hipotiroidismo Cetoacidosis Diabética Coma Hiperosmolar Hiperglicémico Tratornos Hidroelectrolíticos Miotoxicidad Directa Estatinas Fibratos Antirretrovirales Colchicina Infecciones Locales y Sistémicas Miositis Micotoxinas Toxinas Animales |
La contracción muscular finaliza con la normalización de los niveles de calcio en el sarcoplasma, al ser transportados de regreso al RS. Esta movilización sucede a través de una bomba de tipo simporte de Ca2+/Mg2 ATPasa en la membrana plasmática, así como en el retículo sarcoplásmico donde se expresa una Ca2+ ATPasa21. Adicionalmente, las mitocondrias en los miocitos pueden contribuir a la regulación del Ca2+ en el EIC, pues también expresan tanto la bomba an- tiportadora 2Na+/Ca2+ ATPasa como la bomba simportadora Ca2+/Mg2 ATPasa15. Debido a la ATP-dependencia de estos sistemas reguladores en el sarcolema y la mitocondria, el ATP es una molécula esencial en la fisiología muscular de manera paralela al Ca2+, en el inicio, ejecución, finalización de la contracción muscular y estabilización del potencial en reposo. Los requerimientos de ATP son cubiertos por sistemas donadores de fosfato –creatinkinasa-fosfocreatina22– y oxígeno –mioglobina23– al igual que potentes maquinarias enzimáticas para la degradación de ácidos grasos y glucosa, que se expresan en proporciones diferentes en cada grupo
1. Rabdomiólisis por Trauma Físico
Estas causas implican la pérdida de la continuidad del sarcolema debido a la acción de un agente físico externo32. Aunque los primeros casos de RM se describieron a partir de causas traumáticas, actualmente es más frecuente en forma subclínica caracterizada por presentar ligeras elevaciones de CK, usualmente tras accidentes de tránsito33,34. La RM traumática es de particular importancia durante catástrofes naturales, colapsos de edificaciones y escenarios bélicos, donde el Síndrome de Aplastamiento es mucho más común35. Si bien la pérdida de la integridad estructural del sarcolema por sí sola provoca la muerte celular y libera el contenido de los miocitos al torrente sanguíneo, el proceso puede ser acelerado y amplificado por el Ca2+. En efecto, la concentración de Ca2+ en el EIC es 10.000 veces menor que la del EEC, por lo cual la pérdida de la continuidad del sarcolema acarrea un influjo masivo de Ca2+ al EIC según su gradiente electroquímico, activando los mecanismos de muerte celular dependientes de Ca2+ 36.
2. Rabdomiólisis por Agotamiento Energético
Este grupo engloba situaciones en las cuales la demanda energética del miocito no puede ser cubierta, lo cual desencadena la activación de diversos mecanismos de muerte ce- lular6; siendo las más frecuentes aquellas por esfuerzo físico excesivo37. La deficiencia en el aporte de ATP resulta en la disfunción de los transportadores iónicos de membrana, tales como la disfunción de la bomba antiportadora Na+/K+ ATPasa –que en condiciones normales exporta Na+ al EEC e importa K+ al EIC– tras el déficit energético genera un aumento de Na+ en el sarcoplasma que, además de causar edema celular, amplifica la actividad de la bomba antiportadora 2Na+/ Ca2+ ATPasa, elevando la concentración sarcoplásmica de Ca2+ y activando subsecuentemente las cascadas pro-muer- te celular38. Adicionalmente, la bomba simportadora de Ca2+/ Mg2 ATPasa, que en este escenario es incapaz de exportar estos iones de regreso al SS36, representa un mecanismo patogénico de “relevo” tras el fracaso temprano de la Na+/K+ ATPasa y 2Na+/Ca2+ ATPasa.
Notablemente, la RM por esfuerzo físico se caracteriza por ser multifactorial en su patogénesis. Entre las condiciones que participan en este contexto (adicionales a la depleción de ATP) se encuentra la ausencia de acondicionamiento físico, siendo este caso de RM más frecuente en individuos sin antecedentes de actividad física habitual, aunque puede suceder incluso en atletas39. La realización de actividad física en ambientes muy cálidos o húmedos potencia la pérdida de líquidos y electrolitos, fundamentalmente K+, a través de la sudoración. Debido a que el K+ parece ser un importante mediador de la vasodilatación en las arterias irrigadoras de los grandes grupos musculares40, la disminución en su concentración puede atenuar este efecto y agravarse ante la hipovolemia derivada de la sudoración, resultando en un decremento severo del aporte sanguíneo al músculo41.
También parece participar un componente inmunológico, como lo es la elevación de IL-642. Este fenómeno se ha asociado a una mayor activación de linfocitos Th17, con incremento en la secreción de IL-17 e IL-23, ligadas a una mayor concentración sérica de mioglobina, mieloperoxidasa y sRANKL, lo cual sugiere que estos mediadores pudieran promover daño muscular a través de neutrófilos activados tras un ejercicio intenso43. Además, la RM por esfuerzo físico parece precipitarse con más frecuencia posterior a la administración de suplementos dietarios ricos en creatina, cafeína, efedra y sus derivados, entre otros44, aunque su vínculo etiopatogénico con la RM no se ha precisado45. Finalmente, en individuos con anemia o rasgo falciforme, la hipoxemia relativa propia del esfuerzo físico puede precipitar vasooclusión regional, restringiendo la irrigación a grupos musculares46.
Otras causas importantes de RM por agotamiento energético se relacionan con la disregulación de la temperatura corporal, como la Hipertermia Maligna, el Síndrome Neuroléptico Maligno, y el Síndrome Serotoninérgico, que como rasgo común comparten hipertonía muscular y aceleración del metabolismo oxidativo que puede llevar a RM si estas demandas no pueden ser alcanzadas47. Si bien una predisposición genética parece explicar la patogenia de RM en la Hipertermia Maligna –con al menos 6 loci de interés, siendo el más prominente el gen RYR1, codificador del receptor de rianodina48– el vínculo fisiopatológico entre los otros dos trastornos y la RM aún no ha sido esclarecido49. Por otro lado, la hipotermia también se ha relacionado con RM, probablemente debido a vasoconstricción intensa o hipoxia general50.
Por su parte, otras causas relacionadas al agotamiento energético involucran interferencias directas sobre vías metabólicas del miocito, resultando en menor síntesis de ATP, como se ha observado en miopatías metabólicas congénitas –entre las cuales la deficiencia de carnitina palmitoiltransferasa es la más frecuente51– y tras la exposición a agentes disruptores de la cadena respiratoria o desacoplantes de la fosforilación oxidativa52. Por último, cualquier situación que implique hiperactividad neuromuscular, como estatus epiléptico53, estatus asmático54 y agitación neuropsíquica55, pueden asociarse a RM en su curso.
3. Otras causas de Rabdomiólisis
Drogas de Abuso
Este grupo de drogas representa una de las principales causas de RM, y se caracterizan por propiciar distintas condiciones que en conjunto finalizan en el desencadenamiento de RM56. El etanol inhibe el tráfico de Ca2+ hacia el RS, provoca disfunción de las bombas ATPasa y altera el metabolismo de carbohidratos en miocitos57. Adicionalmente, los sujetos alcohólicos suelen poseer algún grado de malnutrición, resultando en menores reservas de glucógeno y ATP que empeora el pronóstico58; así mismo se ha reportado que en sujetos con delirium tremens –que cursan con temblor e hipertonicidad prolongada puede conllevar a RM59. Por su parte la cocaína y adulterantes utilizados en su preparación60 parecen capaces de iniciar un mecanismo miotóxico directo61 y favorecer isquemia en masas musculares a través de su efecto vaso constrictor62. Más allá de esto, la cocaína parece disminuir la expresión de receptores de dopamina, predisponiendo a la aparición de distonía e hipertermia63, este mecanismo es compartido con las anfetaminas, las cuales además se asocian a señalización dopaminérgica y noradrenérgica incrementada57, e hiperactividad neuropsíquica, largos períodos de vigilia y disminución del apetito57, factores que en conjunto restringen el aporte energético y elevan los requerimientos en el tejido muscular64,65. Por último, las fenilciclidinas parecen favorecer la RM sólo a través de su acción como simpatomiméticos, causando mayor gasto de ATP y a menudo asociándose a mayor práctica de actividad física66.
Miotoxinas Directas
Algunas drogas pueden ejercer efectos deletéreos directos sobre el miocito, este es el caso de las estatinas y fibratos, que ocupan el segundo lugar en frecuencia como causas de RM56. Las estatinas inducen la expresión de calmodulina y enzimas proteolíticas, al igual que incrementan la actividad de la vía ubiquitina-proteasoma en los miocitos67. Por otro lado, el mecanismo lesivo de los fibratos es menos claro, aunque parecen generar daño nuclear condicionando sobreexpresión de PPAR-α68. Ambas drogas poseen un efecto aditivo sobre el riesgo de RM, y en general su uso concomitante no se recomienda69. De manera similar, se han reportado casos de RM asociada a antiretrovirales, en particular tenofovir70 y en asociación a estatinas71, cuyos mecanismos subyacentes se desconocen. Otro fármaco miotóxico es la colchicina, cuyo uso crónico se asocia a alteraciones del funcionamiento microtubular y del tráfico de organelos en miocitos y motoneruonas, al igual que la miopatía vacuolar no necrótica72. Se ha observado un potencial efecto micotóxico asociado a RM en los basidiomicetos del género Russula, aunque sus rasgos toxicológicos no han sido dilucidados73. El veneno de ciertas especies de ofidios –específicamente, Bothrops y Crotalus74– incluye fosfolipasa A2 y otras miotoxinas en su composición, que se añaden al efecto traumático de la mordedura75. También se ha observado RM tras pica- duras de abejas y avispas y se asume un efecto miotóxico directo, si bien los mecanismos no han sido precisados76.
Finalmente, se han descrito otras causas de RM pero los mecanismos fisiopatológicos involucrados son incomprendidos. Entre estas se incluyen varios desórdenes electrolíticos, principalmente hipokalemia77 e hipofosfatemia78, que son especialmente peligrosos en el escenario clínico debido a la liberación local de K+ y fosfato proveniente de los miocitos que enmascara el déficit global. Otras alteraciones de esta índole incluyen hipocalcemia, hiponatremia e hipernatremia6. Trastornos endocrinos también se han asociado, como los estados diabéticos cetoacidóticos e hiperosmolares, que parecen agravarse o depender de la hipofosfatemia y otros desórdenes electrolíticos79, entre otras endocrinopatías. También se ha observado en pacientes con trastornos inflamatorios del tejido muscular80; al igual que infecciones virales, –influenza, parainfluenza, coxsackievirus, dengue, herpes simplex, VIH, entre muchos otros81– bacterianas –más frecuentemente tras infecciones del tracto respiratorio por organismos Gram-negativos82, o en pacientes con sepsis83– e incluso parasitarias como Plasmodium84 Leishmania y Toxoplasma, al igual que hongos, principalmente Candida38.
Muerte miocítica
Como señalamos previamente existe una amplia gama de causas responsables de iniciar la RM, sin embargo el mecanismo patogenético coincide con la mayoría, el cual se caracteriza por un incremento en la concentración de calcio iónico citoplasmático85, y una vez que el calcio supera un límite crí- tico, se activa la cascada que conlleva a la destrucción del miocito, ver Figura 2.
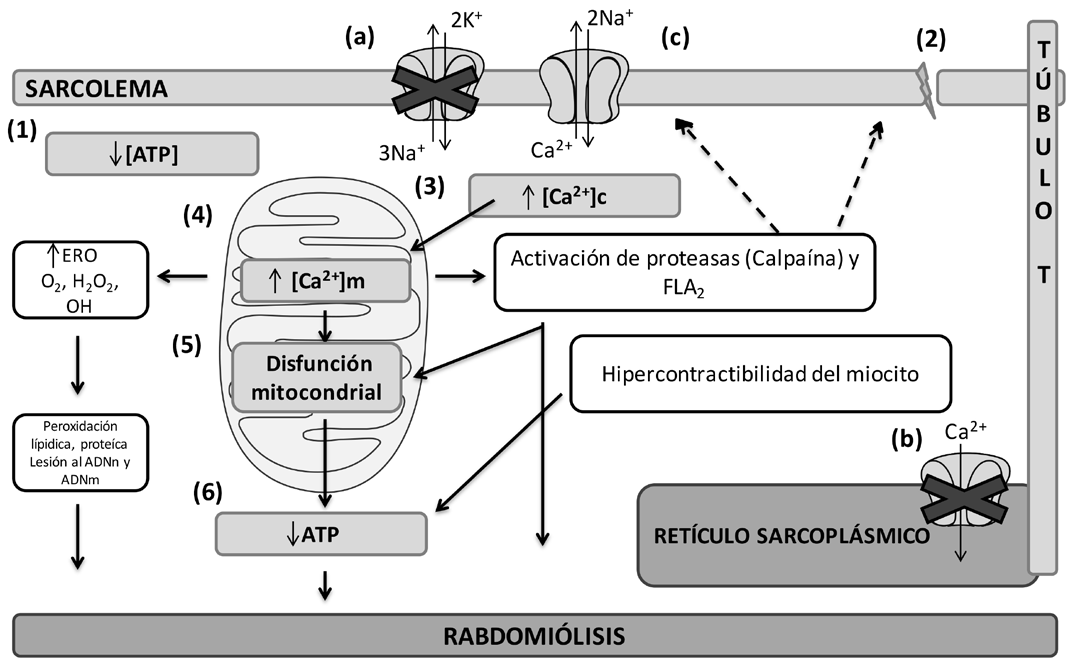
Figura 2. Eventos fisiopatológicos de la rabdomiólisis.
El agotamiento de la producción de ATP (1) y ruptura del sarcolema (2) aumenta la concentración de calcio citoplasmático y mitocondrial (3,4). El aumento de la concentración de este ion da inicio a una cascada de eventos intracelulares e intramitocondriales corriente abajo que finalizan con rabdomiólisis, y posterior liberación de sustancias intramiociticas en el líquido extracelular (5,6).
(a) ATPasa 3Na+/2K+, (b) Ca2+ ATPasa (c) 2Na+/Ca2+ ATPasa [Ca2+]c: Ca2+ citoplasmático, [Ca2+]m: Ca2+ mitocondrial, ERO: especies reactivas de oxígeno, FLA2 : Fosfolipasa A2, ADNn: ADN nuclear, ADNm: ADN mitocondrial. La línea discontinua representa la retroalimentación positiva.
1. Activación de lipasas y proteases
En primera instancia el aumento en la concentración de Ca2+ conlleva a la activación de un grupo de enzimas Ca2+ dependientes, una esterasa, la fosfolipasa A2 (FLA2), y un grupo de proteasas nucleares, entre estas la calpaína1, las cuales degradan los fosfolípidos de las membranas celulares (plasmáticas y mitocondriales) y varios organelos intracelulares86,87. Posterior a la disolución enzimática de los fosfolípidos de membrana, los lisofosfolípidos y ácidos grasos conllevan a daño tóxico directo del sarcolema y otras membranas intracelulares o producen alteración en el funcionamiento adecuado de las proteínas transportadoras, conllevando al flujo de Ca2+ hacia el citoplasma88,89. Además la activación de la calpaína genera la destrucción y pérdida de la desmina y la titina90, la desmina es el principal filamento intermedio en el músculo esquelético, formando uniones entre las líneas Z, miofibrillas y la membrana plasmática, por su lado la titinaregula la integridad estructural de la sarcómera durante los ciclos de contracción y relajación muscular91.
2. Contracción miocítica persistente
Debido a la elevada concentración de Ca2+, el miocito se mantiene en un estado de contracción continuo, lo cual resulta en una mayor reducción del ATP y por ende un agota- miento de las reversas energéticas33.
3. Elevación en el calcio mitocondrial-disfunción mitocondrial
El exceso de Ca2+ citoplasmático genera el paso del ion hacia la mitocondria por gradiente de concentración, este organelo sirve de almacén ante elevación de sus niveles, permitiendo a la célula enfrentarse a la noxa que ocasiono el incremento92. Si la sobrecarga de Ca2+ persiste, se afectan la integridad estructural y funcional de la mitocondria lo cual conlleva a disrupción de la fosforilación oxidativa, y por ende alteración en la producción de ATP. La disminución en la producción de ATP perpetúa la disfunción de los transportadores de Ca2+ en el sarcolema y organelos intracelulares, iniciando así un círculo vicioso con mayor ingreso de Ca2+ y disminución en la producción de ATP93.
4. Producción de radicales libres-estrés oxidativo
Los altos niveles de Ca2+ a nivel mitocondrial conlleva a la producción de especies reactivas de oxígeno (ERO) como súper oxido (O2 ), peróxido de hidrogeno (H2O2), hidroxilo (OH ), exceden la capacidad de unión a proteínas transportadoras y pueden precipitar en el filtrado glomerular104.
La mioglobina es una proteína filtrada libremente por el glomérulo, que entra en la célula tubular epitelial a través de un mecanismo de endocitosis, donde es metabolizada, apareciendo en orina solo cuando se supera el umbral de 1.5 mg/dL, y es macroscópicamente evidente como una orina de color rojiza-marrón cuando los niveles séricos superan los 100 mg/dL101. Los mecanismos implicados, en el deterioro de la filtración glomerular, dependiente de la mioglobina aún no están completamente esclarecidos, se ha propuesto un fenómeno de vasoconstricción renal, lesión tubular directa e
entre otras94. Las ROS posee la capacidad de oxidar varias biomoléculas incluyendo proteínas, lípidos y ácidos nucleicos, favoreciendo una mayor disfunción estructuro-funcional de la célula, debido a la destrucción de los componentes básicos de las membranas y organelos intracelulares, afectando la integridad del sarcolema y membranas intracelulares, así como el funcionamiento de mitocondria y RS95,96. De igual forma este aumento en las ROS, conlleva a mutaciones en el ADN nuclear que produce alteraciones en la disposición organizacional de la célula, y además mutaciones en el ADN mitocondrial que contiene la secuencia codificante de proteínas de la cadena respiratoria, lo cual produce degeneración isquémica y la obstrucción tubular105.
Así mismo, esta molécula se concentra a lo largo de los túbulos renales, un proceso que es favorecido por el agota- miento del volumen -debido a reabsorción agua- y a vaso constricción renal, precipitando al interactuar con la proteína de Tamm-Horsfall o uromodulina, la cual es la glicoproteína más abundante de la vía urinaria, expresada únicamente a nivel de la rama ascendente gruesa del asa de Henle100,106. Además de esto, los altos índices de formación y excreción urinaria de ácido úrico contribuyen a la obstrucción tubular 33 del sistema de transporte de electrones, resultando en menor mediada por los sedimentos de sus cristales, ambos me- producción de ATP97.
5. Muerte de la célula muscular
Por último, el camino que llevará a la muerte del miocito dependerá en parte de la concentración de Ca2+, de manera que niveles esperados de este ion podría desencadenar apoptosis, mediante la activación de factores pro-apoptóticos como citocromo C o factor inductor de apoptosis (AIF) que participan como cofactores de caspasa potenciando la cascada proteolítica apoptótica31. No obstante, este es un proceso que implica altas demandas energéticas para la célula por lo cual solo tomara lugar, si los niveles de ATP son suficientes98. Por otra parte, un ingreso excesivo de este ion a la célula ocasionaría una rápida lisis de la misma mediante inflamación del citoplasma y sus organelos, haciendo que la célula muera y libere su contenido en el espacio extracelular1. Este contenido origina daño a los capilares adyacentes, incluyendo edema local, aumento de la presión intracompartamental y eventualmente isquemia regional, la cual favorece aún más la disminución de los niveles energéticos, destruyendo una mayor cantidad de capilares99. Los leucocitos circulantes se adhieren a los capilares destruidos, se activan y migran a las células musculares en deceso, donde liberan enzimas pro- teolíticas y ROS, agravando aún más la disfunción celular99.
Rabdomiólisis e insuficiencia renal aguda
La insuficiencia renal aguda asociada con mioglobinuria es la complicación más grave de la RM tanto traumática como no traumática, sin embargo su incidencia es difícil de establecer debido a las distintas definiciones y formas clínicas de presentación100, diversos reportes plantean una incidencia entre 13 y 54%56,101-103. En condiciones fisiológicas, la concentración de mioglobina es bastante baja (0-0,003) mg/dL, si existe una lesión que afecte más de 100 gramos de músculo es- quelético, los niveles de mioglobina que se liberan y circulan, canismos se encuentran particularmente favorecidos por la acidificación de la orina. La citotoxicidad celular ocurre principalmente en los túbulos proximales y la obstrucción a nivel de los túbulos distales.
La nefrotoxicidad dependiente de mioglobina depende a su vez de un medio acido urinario, ya que un pH ácido favorece la actividad pseudoperoxidasa, pro-oxidante y la formación de enlaces cruzados en la molécula de mioglobina107. De igual forma esta proteína contiene hierro, en la forma de óxido ferroso (Fe2+), el cual es necesario para ligar el oxígeno molecular, sin embargo este último puede promover la oxidación del Fe2+ a óxido férrico (Fe3+), generando radical hidroxilo, si bien este potencial oxidativo es contrarrestado por moléculas anti-oxidantes, al existir una liberación excesiva de mioglobina se producen ERO y radicales libres capaces de lesionar la célula108, igualmente como resultado de la degradación intratubular de la mioglobina, se libera hierro libre, el cual cataliza la producción de radicales libres y estimula el daño isquémico. Otro aspecto a considerar es que aun cuando existe ausencia de la liberación de hierro libre, el centro hemo de la mioglobina tiene la capacidad por si solo de iniciar peroxidación lipídica y lesión renal109.
Rabdomiólisis en enfermedades crónicas
La RM es una complicación poco común que puede ser observada en trastornos crónicos como la Diabetes Mellitus (DM), no obstante la aparición de esta se encuentra subestimada, debido al carácter subclínico con el que se presenta110. Singhal et al.,111 encontró que más de un 50% (16 de 31) de sus pacientes admitidos con estado hiperosmolar desarrollaron RM, por otro lado Lee et al79 encontró la presencia de RM en aproximadamente un 20% (31 de 149) de pacientes con coma hiperosmolar hiperglicémico no cetónico. El mecanismo a través del que el estado hiperosmolar induce RM, parece involucrar la combinación de distintos factores: por un lado las concentraciones séricas de sodio, la osmolalidad y la hiperglucemia son los factores que precipitan la aparición de RM, aunado a esto, la hipokalemia e hipofosfatemia parecen también favorecer la aparición de RM, estos dos últimos pueden encontrarse enmascarados debido a la liberación de K+ y fosfato por el músculo lesionado110,112.
Igualmente el uso de ciertas drogas en el tratamiento de las alteraciones metabólicas observadas en pacientes con DM parecen estar involucradas en el desarrollo de RM113, la amiodarona inhibe CYP3A4 (isoenzima citocromo P450 que está involucrada en el mecanismo de diversas drogas), además una gran variedad de estatinas como lovastatina, simvastatina o artovastatina, que son metabolizadas primariamente por esta enzima, y cuyo uso monoterápico parece aumentar hasta cuatro veces el riesgo de RM114-117.
Por otra parte el Síndrome de Cushing, que resulta de una exposición crónica y patológica a los glucocorticoides puede conllevar a RM118. La hipertensión se encuentra presente en casi un 80% de los pacientes, aun cuando los mecanismos fisiopatológicos involucrados en este son desconocidos, distintos mecanismos se han propuesto, entre estos la presencia de un estado funcional de exceso mineralocorticoides secundario a la saturación por sustrato de la enzima 11β-hidroxiesteroide deshidrogenasa (11β-HEH), cuya presentación incluye hipokalemia la cual puede propiciar un escenario adecuado para la RM119.
Conclusiones
La rabdomiólisis se trata de un trastorno en el cual existe destrucción de la célula muscular, si bien su etiología es multifactorial, todas estas comparten mecanismos fisiopatológicos que involucran el aumento en la concentración de Ca2+ que resultan en una serie de alteraciones morfofuncionales de la célula miocítica, conllevando a su muerte y liberación de su contenido lo cual resulta altamente tóxico en la circulación plasmática, afectando especialmente la función renal del individuo que la padece.
Es importante pensar en este fenómeno en aquellos individuos que realizan actividades físicas vigorosas en exceso que pueden conllevar a la lesión del músculo esquelético, igualmente, debe considerarse como parte de los fenómenos agravantes en infecciones y enfermedades crónicas, uso de drogas e individuos polimedicados como los pacientes VIH.
Conflictos de interés
Los Autores no tienen conflictos de interés que declarar
Referencias
1. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdo- myolysis: Pathophysiology and diagnosis. Eur J Intern Med. 2007 Mar;18(2):90-100.
2. Rutecki GW, Ognibene AJ, Geib JD. Rhabdomyolysis in antiquity. From ancient descriptions to scientific explication. Pharos Alpha Omega Alpha Honor Med Soc. 1998 Spring;61(2):18-22.
3. Rizzi D, Basile C, Di Maggio A, Sebastio A, Introna F Jr, Rizzi R, et al. Clinical spectrum of accidental hemlock poisoning: neurotoxic manifestations, rhabdomyolysis and acute tubular necrosis. Nephrol Dial Transplant. 1991;6(12):939-43.
4. Bywaters EG, Beall D. Crush injuries with impairment of renal func- tion. 1941. J Am Soc Nephrol. 1998 Feb;9(2):322-32.
5. Mohaupt MG. Rhabdomyolysis. Ther Umsch. 2003 Jul;60(7):391-7.le L. Rhabdomyoysis. Pathophysiology, recognision, and management. Critical Care Nurse 2003; 23: 14-32.
6. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013 Feb 5;17(2):162-
7. Pandy MG, Andriacchi TP. Muscle and joint function in human locomotion. Annu Rev Biomed Eng. 2010 Aug 15;12:401-33.
8. Wingertzahn MA, Ochs RS. Control of calcium in skeletal muscle excitation-contraction coupling: implications for malignant hyperthermia. Mol Genet Metab. 1998 Oct;65(2):113-20.
9. Cooke R. The sliding filament model: 1972-2004. J Gen Physiol. 2004 Jun;123(6):643-56.
10. Hynes TR, Block SM, White BT, Spudich JA. Movement of myosin fragments in vitro: domains involved in force production. Cell. 1987 Mar 27;48(6):953-63.
11. Lehman W, Craig R, Vibertt P. Ca2+-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature. 1994. 368, 65–67.
12. Mazzarello P, Calligaro A, Vannini V, Muscatello U. The sarcoplasmic reticulum: its discovery and rediscovery. Nat Rev Mol Cell Biol. 2003 Jan;4(1):69-74.
13. Guerini D, Coletto L, Carafoli E. Exporting calcium from cells. Cell Calcium. 2005 Sep-Oct;38(3-4):281-9.
14. Mazzarello P, Calligaro A, Vannini V, Muscatello U. The sarcoplasmic reticulum: its discovery and rediscovery. Nat Rev Mol Cell Biol. 2003 Jan;4(1):69-74.
15. Flucher BE. Structural analysis of muscle development: transverse tubules, sarcoplasmic reticulum, and the triad. Dev Biol. 1992 Dec;154(2):245-60.
16. Franzini-Armstrong C, Kenney LJ, Varriano-Marston E. The structure of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study. J Cell Biol. 1987 Jul;105(1):49-56.
17. Araya R, Liberona JL, Cárdenas JC, Riveros N, Estrada M, Powell JA, et al. Dihydropyridine receptors as voltage sensors for a depo- larization-evoked, IP3R-mediated, slow calcium signal in skeletal muscle cells. J Gen Physiol. 2003 Jan;121(1):3-16.
18. Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997 Mar;49(1):1-51.
19. Toyoshima C, Mizutani T. Crystal structure of the calcium pump with a bound ATP analogue. Nature. 2004 Jul 29;430(6999):529-35.
20. Echegaray M, Rivera MA. Role of creatine kinase isoenzymes on muscular and cardiorespiratory endurance: genetic and molecular evidence. Sports Med. 2001;31(13):919-34.
21. Garry DJ, Mammen PP. Molecular insights into the functional role of myoglobin. Adv Exp Med Biol. 2007;618:181-93.
22. Picard M, Hepple RT, Burelle Y. Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am J Physiol Cell Physiol. 2012 Feb 15;302(4):C629-41.
23. Efstratiadis G, Voulgaridou A, Nikiforou D, Kyventidis A, Kourkouni E, Vergoulas G. Rhabdomyolysis updated. Hippokratia. 2007 Jul;11(3):129-37.
24. Maduell F, Navarro V, Cruz MC, Torregrosa E, Garcia D, Simon V, et al. Osteocalcin and myoglobin removal in on-line hemodiafiltration versus low- and high-flux hemodialysis. Am J Kidney Dis. 2002 Sep;40(3):582-9.
25. Ordway GA, Garry DJ. Myoglobin: an essential hemoprotein in striated muscle. J Exp Biol. 2004 Sep;207(Pt 20):3441-6.
26. Grange RW, Meeson A, Chin E, Lau KS, Stull JT, Shelton JM, et al. Functional and molecular adaptations in skeletal muscle of myoglobin-mutant mice. Am J Physiol Cell Physiol. 2001 Nov;281(5):C1487-94.
27. Kanatous SB, Mammen PP. Regulation of myoglobin expression. J Exp Biol. 2010 Aug 15;213(Pt 16):2741-7.
28. Schenkman KA, Marble DR, Burns DH, Feigl EO. Myoglobin oxygen dissociation by multiwavelength spectroscopy. J Appl Physiol (1985). 1997 Jan;82(1):86-92.
29. Rizzuto R, Pinton P, Ferrari D, Chami M, Chami M, Szabadkai G, et al. Calcium and apoptosis: facts and hypotheses. Oncogene.2003; 22: 8619-8627.
30. Lane R, Phillips M. Rhabdomyolysis. BMJ. 2003 Jul 19;327(7407):115-6.
31. Vanholder R, Sever MS, Erek E, Lameire N. Rhabdomyolysis. J Am Soc Nephrol. 2000 Aug;11(8):1553-61.
32. Vanholder R, Sever M, Erek E, Lemeire N. Acute renal failure related to the crush syndrome: towards an era or seismonephrology? Nephrol Dial Transplant. 2000, 15:1517-1521.
33. Kantarci G, Vanholder R, Tuglular S, Akin H, Koc M, Ozeenr C, et al. Acute renal failure due to crush syndrome during Marmara earth- quake. Am J Kidney Dis 2002, 40:682-689.
34. Zhang M. Rhabdomyolysis and its pathogenesis. World J Emerg Med. 2012; 3(1):11-15.
35. George M, Delgaudio A, Salhanick SD. Exertional rhabdomyolysis-when should we start worrying? Case reports and literature review. Pediatr Emerg Care. 2010 Nov;26(11):864-6.
36. Guis S, Mattei JP, Cozzone PJ, Bendahan D. Pathophysiology and clinical presentations of rhabdomyolysis. Joint Bone Spine. 2005 Oct;72(5):382-91.
37. Sharma N, Winpenny H, Heymann T. Exercise-induced rhabdomyolysis: even the fit may suffer. Int J Clin Pract. 1999 Sep;53(6):476-7.
38. Eckman DM, Nelson MT. Potassium ions as vasodilators: role of in- ward rectifier potassium channels. Circ Res. 2001 Feb 2;88(2):132-3.
39. Jain VV, Gupta OP, Jajoo SU, Khiangate B. Hypokalemia induced rhabdomyolysis. Indian J Nephrol. 2011 Jan;21(1):66.
40. Fischer CP. Interleukin-6 in acute exercise and training: what is the biological relevance? Exerc Immunol Rev. 2006;12:6-33.
41. Sugama K, Suzuki K, Yoshitani K, Shiraishi K, Kometani T. IL-17, neutrophil activation and muscle damage following endurance exer- cise. Exerc Immunol Rev. 2012;18:116-27.
42. Burke J, Seda G, Allen D, Knee TS. A case of severe exercise-in- duced rhabdomyolysis associated with a weight-loss dietary supple- ment. Mil Med. 2007 Jun;172(6):656-8.
43. Scroggie DA, Harris M, Sakai L. Rhabdomyolysis associated with nutritional supplement use. J Clin Rheumatol. 2000 Dec;6(6):328-32.
44. Anzalone ML, Green VS, Buja M, Sanchez LA, Harrykissoon RI, Eichner ER. Sickle cell trait and fatal rhabdomyolysis in football training: a case study. Med Sci Sports Exerc. 2010 Jan;42(1):3-7.
45. Bertorini TE. Myoglobinuria, malignant hyperthermia, neuroleptic malignant syndrome and serotonin syndrome. Neurol Clin. 1997 Aug;15(3):649-71.
46. Litman RS, Rosenberg H. Malignant hyperthermia: update on susceptibility testing. JAMA. 2005 Jun 15;293(23):2918-24.
47. Rajapakse S, Abeynaike L, Wickramarathne T. Venlafaxine-associated serotonin syndrome causing severe rhabdomyolysis and acute renal failure in a patient with idiopathic Parkinson disease. J Clin Psychopharmacol. 2010 Oct;30(5):620-2.
48. Bonnor R, Siddiqui M, Ahuja TS. Rhabdomyolysis associated with near-drowning. Am J Med Sci. 1999;318(3):201.
49. Kaneoka H, Uesugi N, Moriguchi A, Hirose S, Takayanagi M, Yama- guchi S, et al. Carnitine palmitoyltransferase II deficiency due to a novel gene variant in a patient with rhabdomyolysis and ARF. Am J Kidney Dis. 2005 Mar;45(3):596-602.
50. Truica CI, Frankel SR. Acute rhabdomyolysis as a complication of cytarabine chemotherapy for acute myeloid leukemia: case report and review of literature. Am J Hematol. 2002 Aug;70(4):320-3.
51. Walker CP, Duddy MJ, Sagar G. Case report: rhabdomyolysis fol- lowing grand mal seizures presenting as a delayed and increasingly dense nephrogram. Clin Radiol. 1993 Feb;47(2):139-40.
52. Barrett SA, Mourani S, Villareal CA, Gonzales JM, Zimmerman JL. Rhabdomyolysis associated with status asthmaticus. Crit Care Med. 1993 Jan;21(1):151-3.
53. Kotbi N, Mahgoub N, Mokonogho J, Young R. Rhabdomyolysis associated with mania in late life. Int J Geriatr Psychiatry. 2009 Dec;24(12):1478-9.
54. Melli G, Chaudhry V, Cornblath DR. Rhabdomyolysis: an evaluation of 475 hospitalized patients. Medicine (Baltimore). 2005;84(6):377.
55. Richards JR. Rhabdomyolysis and drugs of abuse. J Emerg Med. 2000 Jul;19(1):51-6.
56. Haller RG, Knochel JP. Skeletal muscle disease in alcoholism. Med Clin North Am 1984;68:91–103.
57. Pariente EA, Nouel O, Bernuau J, Fraisse F, Degott C, Rueff B. Acute rhabdomyolysis in alcoholic patients. Presse Med. 1983 Feb 12;12(6):339-43.
58. Nolte KB. Rhabdomyolysis associated with cocaine abuse. Hum Pathol. 1991 Nov;22(11):1141-5.
59. Pagala M, Amaladevi B, Azad D, et al. Effect of cocaine on leakage of creatine kinase from isolated fast and slow muscles of rat. Life Sci 1993;52:751– 6.
60. Brody Sl, Wrenn KD, Wilber MW, et al. Predicting the severity of cocaine-associated rhabdomyolysis. Ann Emerg Med 1990;19: 1137–43.
61. Cappon GD, Morford LL, Vorhees CV. Ontogeny of methamphet- amine- induced neurotoxicity and associated hyperthermic response. Brain Res 1997;103:155– 62.
62. Morgan P, Beck JE. The legacy and the paradox: hidden contexts of methamphetamine use in the United States. In: Klee H, ed. Amphetamine misuse: international perspectives on current trends. Amster- dam: Harwood Publishers; 1997:135– 62.
63. Scandling J, Spital A. Amphetamine-associated myoglobinuric renal failure. South Med J. 1982;75:237– 40
64. Cogen FC, Rigg G, Simmons JL, Domino EF. Phencyclidineassoci- ated acute rhabdomyolysis. Ann Intern Med 1978;88:210 –2.
65. Chapman MJ, Carrie A. Mechanisms of statin-induced myopathy: a role for the ubiquitin-proteasome pathway?. Arterioscler Thromb Vasc Biol. 2005 Dec;25(12):2441-4.
66. Maiguma T, Fujisaki K, Itoh Y, Makino K, Teshima D, Takahashi-Yanaga F, et al. Cell specific toxicity of fibrates in human embryonal rhabdomyosarcoma cells. Nauny Schmiedebergs. Arch Pharmacol. 2003 Mar;367(3):289-96
67. Jacob S, Jacob S, Williams C, Deeg M. Simvastatin, Fenofibrate, and Rhabdomyolysis. Diabetes Care 2005 May;28(5):1258.
68. Spiegel LR, Schrier PB, Shah HH. Severe recurrent rhabdomyolysis induced acute kidney injury in a HIV-infected patient on antiretrovi- ral therapy. Ren Fail. 2013 Sep;35(8):1186-90.
69. Burger D, Stroes E, Reiss P. Drug interactions between statins and antiretroviral agents. Curr Opin HIV AIDS. 2008 May;3(3):247-51.
70. Salem C, Sakhri J, Fathallah N, Trimech B, Hmouda H, Kamel B. Colchicine-Induced Rhabdomyolysis and Possible Amiodarone Interaction-Colchicine--Induced Rhabdomyolysis. Pharmacology & Pharmacy 2010;1(2):39-41.
71. Saviuc P, Danel V. New syndromes in mushroom poisoning. Toxicol Rev. 2006;25(3):199-209.
72. Zornetta I, Caccin P, Fernandez J, Lomonte B, Gutierrez JM, Mon- tecucco C. Envenomations by Bothrops and Crotalus Snakes Induce the Release of Mitochondrial Alarmins. PLoS Negl Trop Dis.2012;6(2):e1526.
73. Kini RM. Excitement ahead: structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon. 2003 Dec 15;42(8):827-40.
74. Akdur O, Can S, Afacan G. Rhabdomyolysis Secondary to Bee Sting. Case Rep Emerg Med. 2013;2013:258421.
75. Jain VV, Gupta OP, Jajoo su, Khiangate B. Hypokalemia induced rhabdomyolysis. Indian J Nephrol. 2011 Jan-Mar; 21(1): 66
76. Singhal PC, Kumar A, Desroches L, Gibbons N, Mattana J. Preva- lence and predictors of rhabdomyolysis in patients with hypophosphatemia. Am J Med. 1992;92(5):458.
77. Wang LM, Tsai ST, Ho LT, Hu SC, Lee CH. Rhabdomyolysis in dia- betic emergencies. Diabetes Res Clin Pract. 1994;26(3):209.
78. Kim HW, Choi JR, Jang SJ, Chang YS, Bang BK, Park CW. Recur- rent rhabdomyolysis and myoglobinuric acute renal failure in a pa- tient with polymyositis. Nephrol Dial Transplant. 2005;20(10):2255.
79. Shanmugam S, Seetharaman M. Viral rhabdomyolysis. South Med J. 2008 Dec;101(12):1271-2.
80. Blanco JR, Zabalza M, Salcedo J, Echeverria L, García A, Vallejo M. Rhabdomyolysis of infectious and noninfectious causes. South Med J. 2002;95(5):542.
81. Betrosian A, Thireos E, Kofinas G, Balla M, Papanikolaou M, Georgiadis G. Bacterial sepsis-induced rhabdomyolysis. Intensive Care Med. 1999 May;25(5):469-74.
82. Mishra SK, Pati SS, Mahanta KC, Mohanty S. Rhabdomyolysis in falciparum malaria--a series of twelve cases (five children and seven adults). Trop Doct. 2010 Apr;40(2):87-8.
83. Knochel JP. Mechanisms of rhabdomyolysis. Curr Opin Rheumatol 1993;5:725–31.
84. Vernon LP, Bell JD. Membrane structure, toxins and phospholipase A2 activity. Pharmacol Ther 1992;54:269–95.
85. Nigam S, Schewe T. Phospholipase A(2)s and lipid peroxidation. Bio- chim Biophys Acta 2000;1488:167–81.
86. Zager RA. Rhabdomyolysis and myohemoglobinuric acute renal fail- ure. Kidney Int 1996;49:314–26.
87. Turrens JF, Beconi M, Barilla J, Chavez UB, McCord JM. Mitochon- drial generation of oxygen radicals during reoxygenation of ischemic tissues. Free Radic Res Commun 1991;12–13:681–9.
88. Murphy RM.Calpains. Skeletal muscle function and exercise. Clin Exp Pharmacol Physiol. 2010 Mar;37(3):385-91.
89. Zhang BT, Whitehead NP, Gervasio OL, Reardon TF, Vale M, Fat- kin D, et al. Pathways of Ca²⁺ entry and cytoskeletal damage following eccentric contractions in mouse skeletal muscle. J Appl Physiol (1985). 2012 Jun;112(12):2077-86.
90. Duchen M. Mitochondria and calcium: from cell signalling to cell death. 2000 November. The Journal of physiology, 529,57-68.
91. Poels PJE, Gabreels FJM. Rhabdomyolysis: a review of the literature. Clin Neurol Neurosurg 1993;95:175–92.
92. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love–hate triangle. Am J Physiol Cell Physiol 2004;287:C817–33.
93. Stark G. Functional consequences of oxidative membrane damage. J Membr Biol 2005;205:1–16.
94. DiMauro S, Tanji K, Bonilla E, Pallotti F, Schon EA. Mitochondrial abnormalities in muscle and other aging cells: classification, causes, and effects. Muscle Nerve 2002;26:597–607.
95. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve 2002;25:332–47.
96. Peter ME. Programmed cell death: Apoptosis meets necrosis. Nature 2011;471, 310–312.
97. Le Bras M, Clement MV, Pervaiz S, Brenner C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol Histopathol 2005;20:205–19.
98. Basnayake K, Cockwell P, Hutchison CA. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009 Oct 1;361(14):1411-2; author reply 1412-3.
99. Holt SG, Moore KP. Pathogenesis and treatment of renal dysfunction in rhabdomyolysis. Intensive Care Med 2001;27:803-11.
100. Ward MM. Factors predictive of acute renal failure in rhabdomyoly- sis. Arch Intern Med 1988;148:1553-7.
101. Herráez J, Torracchi A, Antolí A, De la Fuente R, Santos M. Rab- domiólisis. Estudio descriptivo de 449 pacientes. Medicina Clínica 2012;139(6):238-242.
102. Khan FY. Rhabdomyolysis: a review of the literature. Neth J Med. 2009 Oct;67(9):272-83.
103. Zager RA, Gamelin LM. Pathogenetic mechanisms in experimental hemoglobinuric acute renal failure. The American journal of physiol- ogy 1989;256(3):446-455.
104. Padmanabhan S, Graham L, Ferreri NR, Graham D, McBride M, Dominiczak AF. Uromodulin, an Emerging Novel Pathway for Blood Pressure Regulation and Hypertension. Hypertension. 2014 Nov;64(5):918-923.
105. Reeder BJ, Sharpe MA, Kay AD, Kerr M, Moore K, Wilson MT. Toxic- ity of myoglobin and haemoglobin: oxidative stress in patients with rhabdomyolysis and subarachnoid haemorrhage. Biochem Soc Trans. 2002 Aug;30(4):745-8.
106. Zager RA, Foerder CA. Effects of inorganic iron and myoglobin on in vitro proximal tubular lipid peroxidation and cytotoxicity. J Clin In- vest. 1992 Mar;89(3):989-95
107. Holt S, Moore K. Pathogenesis of renal failure in rhabdomyolisis: The role of myoglobin. Exp Nephrol 8:72-76, 2000.
108. Gangopadhyay KK, Ryder RE. Nontraumatic rhabdomyolysis: an unusual complication of diabetic hyperosmolar nonketot- ic (HONK) state. J R Soc Med. 2006 Apr;99(4):200.
109. Singhal PC, Abramovici M, Venkatesan J. Rhabdomyolysis in the hyperosmolar state. Am J Med 1990;88:9–12.
110. Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine 1982;61:141–52.
111. Varughese G, Scarpello J. Non-traumatic rhabdomyolysis: the emerging role of CYP 3A4 in diabetes mellitus. J R Soc Med. Aug 2006; 99(8): 385–386.
112. Law M, Rudnicka AR. Statin safety: a systematic review. Am J Car- diol 2006;97: S52-60.
113. González-Casanova JE. Pertuz Cruz SL, Chávez Vivas M, Rojas-Gómez DM. Influencia de disruptores endocrinos medioambientales sobre la adipogénesis. AVFT – Arch Venez Farmacol Ter. 2018;37(1):164-72.
114. Cano C, Bermúdez V, Sulbarán G, Morales R, Medina M, Amell A, et al. Influencia de la Edad y el Sexo en el Balance Oxidación/Antioxidación. AVFT – Arch Venez Farmacol Ter. 2001;20(1):63-68.
115. ObregónO, Gestne A, Lares M, Castro J, Stulin I, Martínez J, et al. Estatinas y factor de necrosis tumoral alfa. Latinoam Hipertens.2010; 5(1): 6-10.
116. Sharma S, Nieman L. Cushing’s Syndrome: All variants, detection, and treatment. Endocrinol Metab Clin North Am. Jun 2011; 40(2): 379–391.
117. Stewart PM. Tissue-specific Cushing’s syndrome, 11beta-hydroxys- teroid dehydrogenases and the redefinition of corticosteroid hormone action. Eur J Endocrinol. 2003 Sep;149(3):163-8.