Artículos
Efecto de Agalsidasa B sobre el músculo esquelético en pacientes con enfermedad de Fabry Clásica
Effect of Agalsidase B on skeletal muscle in patients with classic Fabry disease
Efecto de Agalsidasa B sobre el músculo esquelético en pacientes con enfermedad de Fabry Clásica
Archivos Venezolanos de Farmacología y Terapéutica, vol. 37, núm. 4, pp. 311-319, 2018
Sociedad Venezolana de Farmacología Clínica y Terapéutica

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-SinDerivar 4.0 Internacional.
Resumen:
La enfermedad de Fabry (EF) es un error innato del catabolismo de los glucoesfingolípidos, ligado al cromosoma X, debido a la deficiencia en la actividad de la α-galactosidasa A lisosomal., ocasionando acumulación de globotriaosilceramida (GL3) en el plasma, y en los lisosomas del endotelio vascular.. Estos pacientes presentan hipoactividad física multifactorial que puede conllevar a una disminución del desarrollo muscular esquelético.
Objetivo: Conocer cuál es el efecto de la terapia de reemplazo enzimático (TRE) con agalsidasa B sobre el músculo esquelético.
Métodos: ensayo clínico autocontrolado de dos pacientes con Enfermedad de Fabry Clásica, a quienes se les tomó muestra de músculo esquelético para microscopía electrónica y estudio histoquímico y bioquímico, antes y después de 6 meses de TRE.
Resultados: se observaron fibras musculares del tipo IIA previo al inicio de la TRE, con conservación de la estructura de la sarcómera, con líneas Z gruesas y rectas, plegamientos del sarcolema, espacios intermiofibrilares ligeramente ampliados, con mitocondrias electrón densas, gotas lipídicas extraídas, tríadas con cisternas terminales edematizadas y glucógeno abundante. Después de 6 meses de TRE, el paciente en mejor estado clínico mostró mayor porcentaje de fibras tipo I y IIA y menor de la tipo IIx; el paciente que recibe diálisis presentó mayor proporción de fibra IIx.
Conclusiones: La EF puede generar hipotrofia del músculo esquelético, probablemente como consecuencia del compromiso mitocondrial y una probable condición de hipoxia muscular crónica no modificable por la terapia con agalsidasa B.
Palabras clave: Ultraestructura, Fabry, Músculo Esquelético, Histoquímica.
Abstract:
Fabry disease (EF) is an inborn error of catabolism of glycosphingolipids , X-linked due to the deficiency in the activity of α - galactosidase A lysosomal., leading to accumulation of globotriaosylceramide (GL3) in plasma and in the lysosomes of vascular endothelial., resulting in an endotheliopathy with a significant systemic manifestations. Patients with Classical Fabry disease have multifactorial physical hypoactivity that can lead to a decrease in skeletal muscle development. Objective: To know what is the effect of enzyme replacement therapy (ERT) with agalsidase B on the biochemical, histochemical and ultrastructural characteristics of skeletal muscle in patients with classic Fabry disease. Methods: A self-controlled clinical trial of two patients with classic Fabry disease, who were took sample of skeletal muscle for electron microscopy and histochemical and biochemical study, before and after 6 months of ERT with agalsidase B. Results: Muscle fibers were observed type IIA, prior to the start of ERT, with conservation of the structure of the sarcomere, with thick Z lines and straight folds of the sarcolemma, slightly extended intermyofibrillar spaces with dense electron mitochondria, extracted lipid droplets and triads with terminal cisternae edematous. In both patients, we observed abundant glycogen. After 6 months of ERT, the observed changes showed little variations; however the patient with a better clinical condition, showed higher percentage of type I and IIA fibers and a decrease on type IIx fibers; the patient receiving dialysis shows highest number of IIx fibers.
Keywords: Ultrastructure, Fabry Disease, Skeletal Muscle, Histochemistry.
Introducción
La enfermedad de Fabry es un error innato del catabolismo de los glucoesfingolípidos, ligado al cromosoma X, debido a la deficiencia en la actividad de la exoglucohidrolasa α-galactosidasa A lisosomal1, lo que ocasiona que se acumulen sistemáticamente globotriaosilceramida (GL3) y glucoesfingolípidos con residuo α-galactosil terminal, en el plasma, y principalmente en los lisosomas del endotelio vascular2, produciendo una endoteliopatía con importantes manifestaciones sistémicas.
La enfermedad de Fabry normalmente se presenta en su forma Clásica caracterizada por acroparestesias, angioqueratomas, hipohidrosis, y opacidad corneal y lenticular, de aparición desde la niñez o la adolescencia. Esta enfermedad afecta principalmente a los varones, hijos de madres portadoras, comprometiendo el funcionamiento de órganos y tejidos tales como la piel, el sistema nervioso central, periférico y vegetativo, el corazón, y los riñones3.
La enfermedad de Fabry, junto con la enfermedad de Batten-Kufs´ (lipofuscinosis ceroide) y la manosidosis, forman un grupo de enfermedades de depósito lisosomal con manifestación asintomática del músculo esquelético, a diferencia de lo que ocurre en la Enfermedad de Pompe, que es otra enfermedad de depósito lisosomal por deficiencia de la α-1,4 glucosidasa, con importante compromiso sintomático del músculo esquelético4,5.
Los pacientes con enfermedad de Fabry Clásica presentan hipoactividad física que puede conllevar a una disminución del desarrollo muscular esquelético, siendo esta hipoactividad de un origen multifactorial, debido entre otras causas a la presencia de lesiones valvulares aórtica y mitral, así como a una falla en la conducción miocárdica, lo que condiciona la aparición de insuficiencia cardíaca, arritmias (que pueden ser complejas y repetitivas) y cardiopatía isquémica; además los pacientes pueden desarrollar hipertrofia ventricular izquierda6, la cual es producto del depósito de glucoesfingolípidos en los miocitos cardíacos y a un incremento absoluto en la masa muscular cardíaca7. En el año de 1977, Time y col, observaron en una biopsia muscular de un paciente con la enfermedad de Fabry que se evidenciaban depósitos de GL3 en las fibras musculares. En las células endoteliales de los capilares musculares, las inclusiones eran más abundantes y pleomórficas8.
La enfermedad de Fabry presenta una alta morbilidad y mortalidad entre los 40 y 50 años de edad. La mortalidad se ha visto reducida gracias a las terapias de sustitución de la función renal (diálisis y/o transplante), y gracias a las técnicas de ingeniería genética que han permitido obtener y purificar, a partir del cultivo de oocito de hámster chino, la enzima agalsidasa B, la cual conserva muchas de las propiedades de la enzima autóloga9, y ha sido autorizada por la FDA en 2003 para ser utilizada en el tratamiento de los pacientes con enfermedad de Fabry10.
Todos estos antecedentes anteriormente mencionados llevaron a plantear como objetivo principal de este trabajo conocer cuál es el efecto de la terapia de reemplazo enzimático (TRE) con agalsidasa B sobre las características bioquímicas, histoquímicas y ultraestructurales del músculo esquelético en pacientes con enfermedad de Fabry Clásica, para tener un mejor conocimiento del compromiso sistémico de esta enfermedad.
Materiales y Métodos
Se llevó a cabo un ensayo clínico autocontrolado en el que se evaluaron dos pacientes de sexo masculino, con edades de 34 y 35 años, respectivamente, con diagnóstico clínico, enzimático e histopatológico renal de Enfermedad de Fabry Clásica, que iban a ser sometidos a terapia de reemplazo enzimático con agalsidasa B (Genzyme Corp; Cambridge, USA), a quienes se les practicó una biopsia de músculo esquelético para su evaluación ultraestructural e histoquímica, antes del inicio de la terapia enzimática y a los 6 meses de terapia enzimática continua. Previamente se había cumplido con los requisitos establecidos en la Declaración de Helsinki (1983), entre ellos la firma del paciente o su representante legal del consentimiento informado. Las características clínicas de ambos pacientes al momento de la primera biopsia se muestran en la tabla 1.
| Características | Paciente 1 CSL | Paciente 2 ASL |
| Edad de aparición de los síntomas (años) | 9 | 9 |
| Edad al DX (años) | 29 | 28 |
| Acroparestesias | Si | Si |
| Angioqueratomas | Si | Si |
| Opacidad corneal | Si | Si |
| Anhidrosis | Si | Si |
| Hipertrofia Ventricular Izquierda (HVI) | Si | Si |
| Arritmia | No | No |
| Dolor abdominal | No | No |
| Falla renal | Si* | No |
| Proteinuria | ** | Si |
| Enfermedad vascular cerebral | No | No |
*Falla renal estadio 5 en hemodiálisis
**Paciente en anuria
Biopsia de Músculo Esquelético
Se procedió a tomar muestra de músculo esquelético en reposo con una aguja de Bergström11. Previamente se desinfectó la piel del paciente y se administró xilocaína al 2% a nivel del tercio medio del vasto lateral del músculo cuadriceps femoral. Luego se realizó una incisión de 5 mm aproximadamente, profundizando hasta llegar al plano muscular; se introdujo la aguja de biopsia con la ventana cerrada, se abrió ésta, y comprimiendo el músculo se produjo el corte del tejido. Al sacar la aguja se comprimió la herida y se cubrió con gasa. La muestra se dividió en tres partes, una para ser usada en el estudio ultraestructural, otra para el estudio histoquímico, y la tercera para la determinación de enzimas. La muestra para microscopía electrónica de transmisión se fijó con glutaraldehído frío al 3%, en buffer fosfato a pH 7,4 y 320 mOsmol a 4.C, siendo postfijada en tetróxido de osmio (OsO4) al 1%. Seguidamente, se deshidrató en una serie de diferentes concentraciones de etanol y se incluyó en la resina Epón. Posteriormente, se hicieron cortes de 1 μm de la muestra incluida, y se tiñeron con azul de toluidina, para poder seleccionar las áreas a estudiar. Las secciones ultrafinas del tejido muscular se recogieron en rejillas de cobre (200 mesh) y se procedió a contrastarlas con acetato de uranilo y citrato de plomo. Por último, dichos cortes se observaron con un microscopio electrónico de transmisión Hitachi-H 500, operado a 100 KV12.
Histoquímica
Para el análisis histoquímico, cada muestra muscular se embebió en OCT (optimal cutting temperature, Tissue Tek II) y se congeló en isopentano, el cual había sido enfriado hasta su punto de fusión por nitrógeno líquido, el cual lo provee de una cubierta protectora que permite prevenir los daños mecánicos durante su manipulación y su almacenamiento; por otra parte sirve de soporte al realizar los cortes en el criostato. Se realizaron secciones transversales seriadas de 10 μm, que se cortaron en el criostato a -20°C, posteriormente se montaron en cubreobjetos para llevar a cabo las siguientes reacciones histoquímicas: la reacción Adenosina Trifosfatasa (ATPasa) miofibrilar, previa incubación ácida (pH: 4,37, 4,6 y 4,8 y alcalina pH: 10,3) según la técnica de Brooke y Kaiser (1970)13; la reacción de NADH-diaforasa, según la técnica de Novikoff, Shin y Drucker (1961)14, y la α-Glicerol fosfato deshidrogenasa, según Wattenberg y Leong (1960)15. Para visualizar los capilares se usó la reacción de α-amilas-PAS de Andersen (1975)16. A las muestras de ambos pacientes se les realizó además la coloración convencional de hematoxilina-eosina.
Bioquímica
La tercera parte de la muestra se congeló directamente en nitrógeno líquido y se guardó en un congelador a -80ºC hasta su procesamiento. La muestra fue homogeneizada en buffer frío de fosfato de potasio y se determinaron las enzimas oxidativas citrato sintetasa del ciclo de Krebs, la 3-hidroxiacil CoA deshidrogenasa (HAD), de la β-hidroxilación de los ácidos grasos y la enzima glicolítica Lactato deshidrogenasa, por métodos fluorométricos (Lowry y Passonneau, 1972)17. Los resultados se expresaron en μmol/min/g de peso húmedo del tejido.
Las biopsias del músculo se realizaron antes y seis meses después de iniciada la terapia de reemplazo enzimático continua.
Terapia de Reemplazo Enzimático (TRE)
Para la aplicación de la TRE se siguieron las normas de tratamiento establecidas por Genzyme Corp. (Cambridge, Estados Unidos de América): en administrar la enzima Agalsidasa B en una dosis de 1 mg/kg de peso corporal cada 15 días, por vía endovenosa, en un tiempo de infusión de 3 a 4 horas. Una hora antes del tratamiento, los pacientes eran pre-medicados con acetaminofen 1 gr vía oral, y clorfeniramina 8 mg vía oral, para evita reacciones secundarias o efectos adversos de la terapia enzimática
Resultados
En los dos pacientes evaluados, el estudio ultraestructural a nivel del músculo esquelético, mostró fibras musculares del tipo IIA (glucolítico-oxidativas) (micrografías No. 1, 2) y previo al inicio de la TRE, se observó conservación de la estructura de la sarcómera, con líneas Z gruesas y rectas y con plegamientos del sarcolema (micrografía No. 1). Los espacios intermiofibrilares se observaron ligeramente ampliados, con mitocondrias electrón densas, gotas lipídicas extraídas y tríadas con cisternas terminales edematizadas. De manera similar que en la micrografía No. 1, en la 2 se aprecia uno de los espacios intermiofibrilares ampliado. Se localizan también mitocondrias y tríadas edematizadas, así como gotas extraídas de lípidos. El glucógeno es abundante en ambos casos (micrografía 1 y 2).
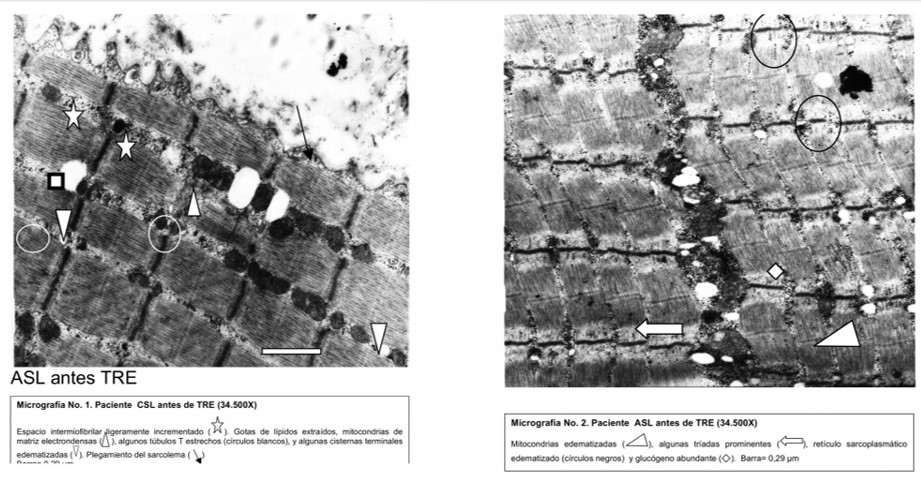
micrografía 1 y 2
Después de los 6 meses de tratamiento (Micrografías 3 y 4), estos cambios no mostraron variaciones de consideración; sin embargo en el paciente ASL, la ultraestructura del músculo esquelético mostró imágenes compatibles con áreas de atrofia de las miofibrillas, con pérdida del aparato contráctil. Este mismo paciente mostró incremento del espacio subsarcolémico con núcleos agrupados de contornos irregulares. La membrana externa de la envoltura nuclear del núcleo central se separó formándose una vacuola con restos de membrana. Además, presentó un sistema sarcotubular fragmentado, mitocondrias degeneradas y miofibrillas supercontraídas con sarcómeros que no muestran la línea M y la banda H (micrografía No. 5).

Micrografías 3

Micrografías 4
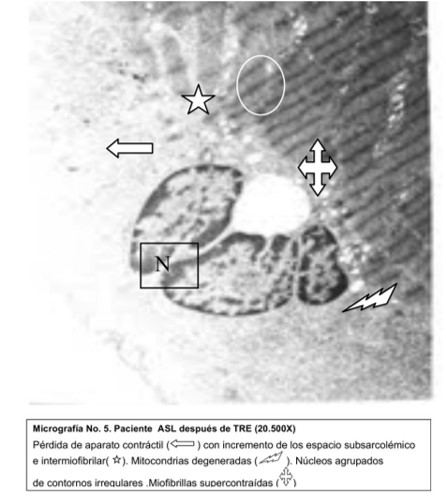
micrografía No. 5
En relación a los resultados histoquímicos de la primera biopsia, se observó que el paciente ASL, quien no tiene falla renal, posee un mayor porcentaje de fibras tipo I (Tabla 2) con respecto al paciente CSL, quien está en hemodiálisis, En el paciente CSL se produjo un aumento importante de las fibras tipo IIx en la segunda biopsia muscular, manteniendo casi constante el porcentaje de fibras tipo I. En el paciente ASL, posterior a la administración de la terapia enzimática, se observó una disminución del porcentaje de las fibras tipo IIx, a favor del aumento de las fibras tipo IIa.
En cuanto a los capilares, en el paciente CSL se encontraron de aspecto normal, sin embargo a los 6 meses se vió la presencia de capilares engrosados y ocluidos.
El paciente ASL presentó presencia de capilares de aspecto anormal tanto en la primera como en la segunda biopsia. En la tinción de hematoxilina-eosina no se observó ninguna anormalidad antes ni después del TRE.
| Paciente CSLPre-TRE | Paciente CSLPost-TRE | Paciente ASLPre-TRE | Paciente ASLPost-TRE | |
| ATPasa | Fibras tipo I 42% Fibras tipo IIa 20% Fibras tipo IIx 38% | Fibras tipo I 40% Fibras tipo IIa 10% Fibras tipo IIx 50% | Fibras tipo I 58% Fibras tipo IIa 21% Fibras tipo IIx:21% | Fibras tipo I 51% Fibras tipo IIa 32% Fibras tipo IIx 17% |
| PAS | Capilares de aspecto normal Índice capilar fibra 1.29. Densidad capilar 326 cap/mm2 | Capilares de aspecto anormal | Capilares de aspecto anormal (algunos ocluidos) | Capilares de aspecto anormal |
En la Figura 1, se muestran los niveles de enzimas musculares en cada paciente antes y después de la TRE; el paciente CSL presentó descenso de un 23,55% de la citrato sintetasa, y de 31.68% de la β hidroxiacil CoA deshidrogenasa, mientras que ASL presentó aumento de ambas enzimas en 16,90% de la citrato sintetasa y 25,06% de la β hidroxiacil CoA deshidrogenasa. En CSL, la actividad enzimática del citrato sintetasa registrada al final del tratamiento es prácticamente igual a ASL al inicio de la terapia. La actividad de la enzima lactato deshidrogenasa es mayor en el paciente CSL, en 25,58% pre tratamiento y en 22,43% posterior al tratamiento con respecto a ASL, aunque en cada paciente no se observaron variaciones importantes antes y después de la TRE.
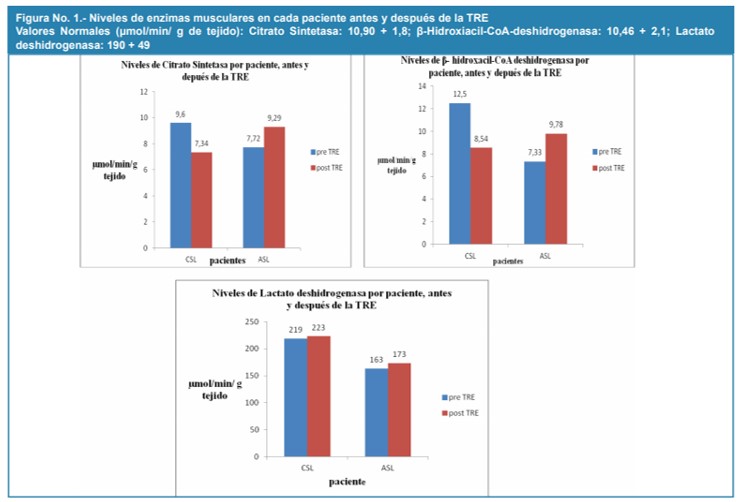
Figura 1
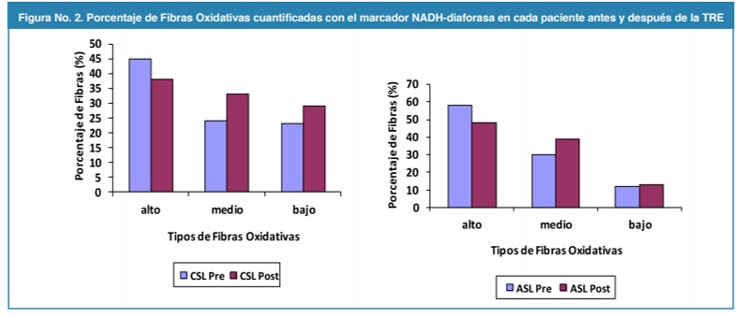
Figura No. 2.
Porcentaje de Fibras Oxidativas cuantificadas con el marcador NADH-diaforasa en cada paciente antes y después de la TRE
La proporción de fibras con alta capacidad oxidativa fue mayor en ASL (58%) que en CSL (45%). Sin embargo en ambos pacientes disminuyó ligeramente durante el TRE, observándose un pequeño aumento de las fibras con capacidad oxidativa media (Fig. 3). Por otra parte CSL mostró una mayor proporción de fibras con alta capacidad glicolítica (60%) comparado con ASL (35%); después del TRE Los cambios fueron moderados notándose en ambos pacientes un aumento de la proporción de fibras con mediana capacidad glicolítica.
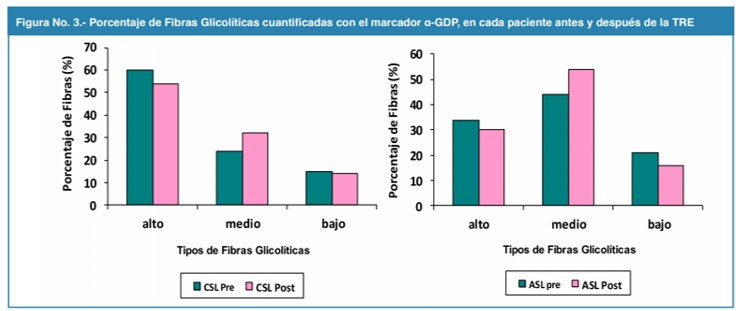
Fig. 3
Porcentaje de Fibras Glicolíticas cuantificadas con el marcador α-GDP, en cada paciente antes y después de la TRE
Discusión
En los pacientes con Enfermedad de Fabry, el compromiso muscular con frecuencia se traduce en fatiga y dolor proximal de los miembros, circunstancias que no están completamente esclarecidas, porque estos síntomas generalmente se consideran como una consecuencia de la insuficiencia cardíaca y/o relacionada con el compromiso de los nervios periféricos. En estos pacientes se describe la presencia de calambres en la fase temprana de le enfermedad, mialgias, fatiga, astenia, y compromiso motor en las actividades diarias, comprometiendo la calidad de vida, en las fases avanzadas de la enfermedad18.
Es importante señalar que la microscopía electrónica es una herramienta muy útil en la evaluación de cualquier patología. En el año de 1995, Uchino y col, observaron en el músculo cardíaco de una mujer de 52 años de edad, portadora de la enfermedad de Fabry, un patrón de mosaico en fibras de apariencia normal y fibras hipertróficas que contenían un número de cuerpos laminados, tipo anillo, además de una amplia acumulación de abundantes cuerpos laminados en las miofibrillas, asociadas con un incremento de los gránulos de glucógeno y de las vacuolas autofágicas19.
En este estudio se pudo observar que en los cortes para microscopía electrónica del músculo esquelético de los pacientes evaluados, se conserva la arquitectura de la sarcómera, por lo que la TRE no logró establecer cambios significativos a nivel ultraestructural, a pesar de que uno de ellos estaba en tratamiento hemodialítico mientras estaba recibiendo la TRE. Después de recibir la TRE, en ambos pacientes se observaron signos llamativos de atrofia muscular, debido al estrechamiento de las miofibrillas y al plegamiento de la membrana del sarcolema, con abundantes gránulos de glucógeno, los cuales se hicieron más evidentes en el paciente con las siglas CSL (Micrografías 3 y 5).
En una variedad de estados patológicos, tales como diabetes mellitus, hiperparatiroidismo, distrofia muscular, entre otros, se ha observado degeneración y atrofia de las miofibrillas, afectando al músculo esquelético. Las miofibrillas en proceso de degeneración muestran una variedad de cambios ultraestructurales, los cuales no son patognomónicos para una enfermedad específica. En lesiones a corto y mediano plazo, existe una disminución del ancho de las miofibrillas debido a la pérdida de los miofilamentos periféricos, pero se mantiene la estructura normal del sarcómero. Los cambios más severos incluyen la destrucción y pérdida de las miofibrillas a lo largo de una o más sarcómeras, y varios cambios en la línea Z. Como consecuencia de ello, existe abundancia relativa del sarcoplasma, y aparecen estructuras prominentes como mitocondrias, tríadas, ambas de aspecto alterado e incluso degeneradas, y partículas de glucógeno, que persisten por tiempo. La pérdida de las miofibrillas, lleva a la disminución de contenido de la fibra muscular, por lo que su superficie se hace ondulada y marcadamente irregular20. De acuerdo a lo anteriormente mencionado, se puede considerar que el factor condicionante de los cambios observados en los músculos esqueléticos de los pacientes evaluados, podría estar relacionado con la presencia de hipoxia tisular crónica como consecuencia del compromiso endotelial. En el caso del paciente CSL, estos cambios son más pronunciados probablemente por la presencia de atrofia neurogénica del músculo esquelético, como se describe en la fase 5 o terminal de la falla renal crónica21. El edema mitocondrial, junto con el edema tisular, constituye un signo que los patólogos lo llaman "tumefacción turbia" que describe la acumulación de agua dentro de la célula sujeta a stress tóxico e involucra una falla de los mecanismos de control osmótico de la célula, y es la primera causa de muerte celular. Parece que el edema mitocondrial lleva a la supresión de la producción de ATP, lo cual provoca falla en la bomba de Na+ dependiente de ATP, ocasionando el edema de los compartimientos celulares. La variedad más común de edema es la debida al compromiso de la matriz de la cámara interna de la mitocondria22.
El sistema lisosomal está pobremente desarrollado en el músculo esquelético normal. No obstante, en cultivos celulares con mioblastos se ha podido demostrar que las células musculares poseen un gran potencial para formar lisosomas4. Sin embargo, Time y col (1977)8 mostraron que el empleo de una biopsia muscular, en la enfermedad de Fabry, evidenciaba depósitos de esos organelos en las fibras musculares. En las células endoteliales de los capilares musculares, las organelas eran más abundantes y pleomórficas y las células musculares satélites estaban esparcidas. En los pacientes evaluados no se observaron en los cortes de músculo esquelético los típicos depósitos de GL3, con aspecto de cebra o en ruedas de cebolla, que caracteriza a la enfermedad de Fabry.
La actividad de la adenosintrifosfatasa (ATP-asa), con preincubación a diferentes pH permite la clasificación de las fibras musculares en fibras tipo I, IIa y IIx. Las fibras tipo I tienen un alto contenido en enzimas oxidativas como la citrato sintetasa, la succinato deshidrogenasa y la β-hidroxacil-CoA-deshidrogenasa, y bajo contenido en enzimas glicolíticas como la fosforilasa, la glícerofosfato deshidrogenasa y la lactato deshidrogenasa; por el contrario las fibras IIx tienen mayor contenido de enzimas glicolíticas y bajo de las oxidativas; las fibras IIa tienen niveles intermedios tanto de enzimas oxidativas como de las glicolíticas. Ciertas condiciones fisiológicas y patológicas permiten variar estos niveles de enzimas, mientras que los cambios en los niveles de la ATP-asa son más difíciles y por lo tanto los cambios de tipo de fibra (al menos de I a II y viceversa) suelen ser limitados, mientras que los cambios entre IIx y IIa acompañan a los cambios en capacidad oxidativa y glicolítica. En nuestros pacientes después del TRE, en el caso de CSL, que estaba en peores condiciones clínicas, hubo un aumento marcado de fibras IIx con disminución de IIa, mientras que en ASL hubo aumento de las fibras IIa.
Los resultados obtenidos con la ATPasa miofibrilar están en concordancia con los observado en relación a la diaforasa de nicotinamida adenina dinucléotido o NADH-diaforasa, enzima que cataliza el proceso de óxido-reducción en el metabolismo aeróbico, los porcentajes de fibras con alta expresión de esta enzima están más elevados en el paciente ASL que en CSL teniendo a su vez, menor porcentaje de fibras de baja expresión de esta enzima. En el caso de la expresión de la α-glicerofosfato deshidrogenada (α-GDP), la cual es una enzima glicolítica, su mayor expresión indica un mayor metabolismo anaeróbico. En el paciente CSL las fibras con alta expresión de dicha enzima están por encima del 50%, y en el paciente ASL las fibras con un nivel mediano de esta enzima son las que predominan. Se puede establecer entonces que existe relación entre las diferentes determinaciones enzimáticas y la expresión del tipo de fibra muscular esquelética, en el paciente CSL existe mayor número de fibras tipo IIx, una menor expresión de la NADH-diaforasa, que es un marcador oxidativo y una mayor expresión de la α-GDP glicolítica. Situación contraria se observa en ASL, quien probablemente por no tener falla renal presenta menor compromiso sistémico (Figura 2 y 3). En los pacientes urémicos se establece una atrofia neurogénica de la musculatura esquelética, predominando en las fibras tipo II, cuyo factor patogénico principal es una alteración en el metabolismo de calcio23. La miopatía urémica está relacionada en la mayoría de los casos con el hiperparatiroidismo secundario, y sus características se sobreponen a la miopatía de la osteomalacia, pudiendo observarse atrofia de las fibras tipo I y tipo II, o de tipo II solamente24. Es posible que la anemia crónica producto de la falla renal estadio 5 en el paciente CSL, favorezca la disminución del porcentaje de fibras tipo I, debido a su menor actividad física por hipoxia tisular y en consecuencia reducción del metabolismo oxidativo. Este aspecto debe ser tomado en cuenta, ya que el paciente CSL recibe tratamiento sustitutivo de la función renal con hemodiálisis, y en él predominan las fibras tipo II (en un 60%)
Todo lo anterior se confirma con la expresión de las enzimas citrato sintetasa, β-hidroxi-deshidrogenasa y lactato deshidrogenasa (Figura 1). La actividad de la enzima citrato sintetasa localizada en la matriz mitocondrial genera: 1.- equivalentes de reducción citosólica para la biosíntesis reductora; 2.- carbono para procesos biosintéticos citosólicos: ácidos grasos, esteroles; 3.- regulación de otros procesos metabólicos, por ejemplo: inhibición de la fosfofructoquinasa, y estimulación de la acetil CoA carboxilasa; y, 4.- a nivel del ciclo de Krebs, generación de energía, en forma de ATP25. La enzima β-hidroxi-deshidrogenasa constituye una de las enzimas del sistema de β oxidación de los ácidos grasos, y se localiza en las membranas internas y en la matriz de las mitocondrias de células animales26. El último paso de la vía glicolítica es una reacción de oxidoreducción catalizada por la lactato deshidrogenada; en ésta reacción se reduce el piruvato a lactato, mientras que el NADH se oxida dando NADH+. Es la única reacción del organismo que permite la síntesis de lactato26.
De acuerdo a todo lo anteriormente mencionado, se puede afirmar que en el paciente CSL existe una alta actividad anaeróbica del músculo esquelético que se mantiene a pesar de la terapia enzimática para su enfermedad de base, muy superior a la observada en el paciente ASL. Es importante recordar que el paciente CSL se encuentra en tratamiento sustitutivo de la función renal con hemodiálisis, y como complicación de ello presentaba para el momento de la toma de la biopsia, signos clínicos y paraclínicos de hiperparatiroidismo secundario. La limitación para el ejercicio observada en pacientes con enfermedad renal crónica se debe a una baja conductancia de oxígeno a nivel del músculo esquelético, y no a una limitada capacidad oxidativa mitocondrial27. En consecuencia el factor principal que determina la capacidad funcional del músculo esquelético, en los pacientes con enfermedad renal crónica, es la perfusión sanguínea, regulada por la integridad de los vasos sanguíneos, más que un compromiso en la estructura celular y subcelular de la fibra muscular estriada.
En la actualidad no se dispone de suficiente información sobre el efecto de la TRE con agalsidasa B sobre la estructura y efectos bioquímicos en el músculo esquelético. No obstante de acuerdo a los resultados obtenidos, en los pacientes evaluados se redujo en 10% el porcentaje de fibras con capacidad oxidativa alta, las fibras con capacidad oxidativa media incrementa entre 7 y 9%. En ambos pacientes se observó un incremento del 8 a 9% de las fibras glicolíticas medias.
En los individuos normales las fibras con más capacidad oxidativa son las tipo I, seguidas por las tipo IIa, teniendo las IIx muy baja capacidad oxidativa. Lo contrario aplica para la capacidad glicolítica. Sin embargo, los cambios enzimáticos suelen ser posibles sobre todo por el estado de actividad de la persona; en cambio las modificaciones en la proporción de fibras entre el tipo I y II están más bien relacionadas con procesos patológicos: en una serie de enfermedades crónicas existe generalmente un aumento de las fibras tipo II a expensas de las de tipo I. Los cambios entre los tipo IIa y IIx son más flexibles y están ligados a la actividad del sujeto. Mientras se es más sedentario, más disminuye la proporción de las fibras IIa y aumenta la de las fibras IIX. La determinación bioquímica de las enzimas oxidativas y glicolíticas es más exacta que la apreciación histoquímica, la cual es más subjetiva, y tiene valor cuando un cambio sobrepasa el 10%. En los pacientes estudiados existe una buena relación entre los resultados bioquímicos y los tipos de fibra aunque también hay coincidencias en relación con las capacidades oxidativa y glicolítica. Así CSL tiene una exagerada proporción de fibras IIx que coincide con una alta capacidad glicolítica; la proporción de fibras tipo IIx se magnificó después del tratamiento, junto con la disminución de las enzimas oxidativas citrato sintetasa y β-hidroxi-CoA deshidrogenasa, haciendo pensar que la progresión de la enfermedad en un paciente que está en diálisis y con la complicación de hipoparatiroidismo, limitó los beneficios del tratamiento. En cambio en ASL se produjo un aumento de las fibras tipo IIa e igualmente aumentaron las enzimas oxidativas, lo cual hace pensar en un efecto terapéutico positivo. Si bien la deshidrogenasa láctica (LDH) no se modificó en ninguno de los dos pacientes, su nivel muestra una tendencia aumentada sobre lo normal en CSL.
Como el estudio se realizó con sólo dos pacientes y considerando que se trata de una enfermedad rara, no se pudo establecer diferencias estadísticamente significativas en relación a las características histoquímicas observadas antes y después TRE, no obstante se puede inferir que la enfermedad de Fabry puede generar hipotrofia del músculo esquelético, probablemente como consecuencia del compromiso mitocondrial y una probable condición de hipoxia muscular crónica producto del daño endotelial propio de esta patología. Los resultados del análisis histoquímico en el paciente CSL parecen estar muy influenciados por el efecto que en el músculo esquelético tiene la enfermedad renal crónica estadío 5, no obstante pareciera que existe un efecto beneficioso de la TRE con agalsidasa B, es por esta razón que se deberían evaluar un mayor número de pacientes en esta línea, para poder concluir su efecto beneficioso en el músculo esquelético de pacientes con Enfermedad de Fabry sin enfermedad renal avanzada.
Referencias
1. –Eng CM, Banikazemi M, Gordon R, Golman M, Phelps R, Kim L, Gass A, Winston J, Dikman S, Fallon J. A Phase ½ Clinical Trial of Enzyme Replacement in Fabry Disease:Pharmacokinetic, Substrate Clearence, and Safety Studies. Am. J. Hum.Genet. 2001; 68:711-722
2.- Miyamura N, Araki E, Matsoda K, Yoshimura R, Furukawa N, Tsuruzoe K, Shirotano T, Kishikawa H, Yamaguchi K and Shichiri M. A Carboxyterminal Truncation of Human α-Galactosidase A in a Heterozygous Female with Fabry Disease and Modification of the Enzymatic Activity by the Carboxy-terminal Domain. Am. Soc. Clin. Invest. 1996; 98 (8): 1809-1817.
3.-Villalobos J, Colina V L. Enfermedad de Fabry: Desde el Aminoácido a la Clínica. VITAE Academia Biomédica Digital. 2008 (36) Julio-Diciembre.
4.- Engel AG. The Scientific basis of Myology En: Engel AG and Banker BQ (eds). Myology Basic and Clinical. New York. McGraw-Hill 1986: 710-711.
5.- Carpenter S and Karpati G. Lysosomal storage in human skeletal muscle. Hum Pathol.1986; 17 (7):683-703.
6.- Warnock DG. Renal aspects of Fabry Disease. Nephrology. 2003; 1(6):802-807.
7.- Kai-Chung Tse, Kwok-Wah Chan, Pui-Chi Tin V, Pok-Siu Yip, Tang S, Fu-Keung Li, Yiu-Wing Ho, Kar-Neng Lai and Tak-Mao Chan. Clinical features and genetic analysis of a Chinese kindred with Fabry´s disease. Nephrol Dial Transplant (2003); 18:182-186
8.- Time FM, Fardeau M and Lenoir G. Ultrastructure of muscle and sensory nerve in Fabry´s disease. Acta Neuropathol. 1977;38(3):187-194.
9.- X Ni, M Canuel, and C.R Morales. The sorting and trafficking of lysosomal proteins. Histol Histopathol. 2006;21:899-913
10.- First treatment for Fabry's disease. FDA Consumer. JulyAugust.2003. FindArticles.com.24Mar2009.http://findarticles.com/p/articles/mi_m1370/is_4_37/ai_106225272
11.- Bergstrom J. Muscle electrolytes in man. Scand J Clin Lab Invest 1962: 68 (Suppl): 1–110.
12.- Villalobos J, Finol H, Febres C, Pulido M, Márquez A, Colina V L. Características Ultraestructurales del Músculo Esquelético en Pacientes con Proteinuria Glomerular. Acta Cient Venez. 2006;56(1):8-14.
13.- Brooke MH, Kaiser KK. Muscle fiber types. How many and what kind? Arch Neurol 1970;23: 369–379.
14.- Novikoff AB, Shin WY, and Drucker J. Mitochondrial localization of oxidative enzymes: Staining results with two tetrazolium salts JCB (1961);9(1): 47-61
15.- Wattenberg LW, Leong JL. Effects of Coenzyme Q10 and menadione on succinic deshidrogenase activity as measured by tetrazolium salt reduction. J Histochem Cytochem, 1960; 8(4): 296-303
16.- Andersen P. Capillary density in skeletal muscles of man. Acata Physiol Scand 1975;95:203-205
17.- Lowry OH and Passonneau JV. A Flexible System of Enzymatic Analysis. New York. Academic Press, 1973; pp1.
18.- Chimeti C, Padua L, Pazzaglia C, Morgante E, Centurion C, Antuzzi D, Russo MA and Frustaci A. Cardiac and skeletal myopathy in Fabry disease: a clinicopathologic correlative study. Hum Pathol. 2012; 43(9): 1444–1452
19.- Uchino M, Uyama E, Kawano H, Hokamaki J, Kugiyama K, Murakami Y, et al. A histochemical and electron study of skeletal and cardiac muscle from a Fabry disease patients and carrieir. Acta Neuropathol 1995; 90 (3):334-338
20.- Ghadially F. Ultrastructural pathology of the cell and matriz. Tomos 1 y 2. 4ta edición. Butterworth-Heinemann, Boston 1977, pp 246-254
21.- Breadly JR, Anderson JR, Evans DB, Cowley AJ,. Impaired nutritive skeletal muscule blood flor in patients with chronic renal failure. Clin. Sci (Colch) 1990; 79 (3):239-45.
22.- Robbins SL. Patología Estructural y Funcional. En Bases Patológicas de la enfermedad, 1th eds. México D.F.: Editorial Interamericana 1975, 177-184
23.- Stein G, Schmidt A, Sperschneider H, Keil E, Michael R, Hedwig R, Funfstruck R, Gassel M. Morphometric and histochemical studies of the skeletal muscles of patients with chronic renal failure and dialysis patients. Z Urol Nephrol.1986;79(19):559-567.
24.- Carrasco-Sánchez FJ, López- Domínguez JM, Casado-Chocan JL, Pérez -Gutiérrez S, Onaidia-Rico JM, Borrero-Martin JJ, Blanco-Ollero A. Uremia myopathy. Rev Neurol 2000;30(12):1154-1156
25.- Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 4th Ed. New York: W. H. Freeman, 2005
26.- Conn E, Stumpf P. Bioquímica Fundamental. 3era edi. México: Editorial Limusa; 1980; pp 330
27.- Sala E, Noyszewski EA, Campistol JM, Marrades RM, Drena S, Torregrossa JV, Beer JS, Wagner PD, Roca J. Impaired muscle oxygen transfer in patients with chronic renal failure. Am J Physiol Regul Integr Comp Physiol. 2001;280(4):R1240-1248