Artículos
Tumor mesenquimal variante no fosfatúrica: a propósito de un caso
Non-phosphaturic variant mesenchymal tumor: A case report
 Roberto José Añez Ramos
Laura González Fernández
Diego Muñoz Moreno
María Miguélez González
Noemí Brox Torrecilla
Juan Carlos Percovich
Roberto José Añez Ramos
Laura González Fernández
Diego Muñoz Moreno
María Miguélez González
Noemí Brox Torrecilla
Juan Carlos Percovich
Tumor mesenquimal variante no fosfatúrica: a propósito de un caso
Archivos Venezolanos de Farmacología y Terapéutica, vol. 40, núm. 5, pp. 524-526, 2021
Sociedad Venezolana de Farmacología Clínica y Terapéutica

Esta obra está bajo una Licencia Creative Commons Atribución-SinDerivar 4.0 Internacional.
Recepción: Febrero , 28, 2021
Aprobación: Marzo , 15, 2021
Publicación: Agosto , 10, 2021
Resumen: Los tumores mesenquimales inductores de osteomalacia (TIO) suelen ser benignos, pequeños, de crecimiento lento. Los TIO tienen una producción anormal del factor de crecimiento fibroblástico 23 (FGF-23) que ocasiona la inhibición de la reabsorción de fosfato en el túbulo contorneado proximal y la inactivación de la 1α-hidroxilasa con la consecuente hipofosfatemia, fosfaturia y alteración del metabolismo de la vitamina D que conlleva a la aparición de osteomalacia adquirida. Recientemente se ha descrito la variante no fosfatúrica. En este caso clínico se presenta un paciente con un tumor mesenquimal no fosfatúrico cuyo diagnóstico fue complicado debido a los síntomas iniciales inespecíficos.
Palabras clave: tumor mesenquimal, fosfaturia, osteomalacia, FGF-23, diagnóstico.
Abstract: Osteomalacia-inducing tumors (OIT) are usually benign, small and slow growing. OIT have an abnormal production of fibroblast growth factor 23 (FGF-23) which causes the inhibition of phosphate reabsorption in the proximal convoluted tubule and the inactivation of 1α-hydroxylase with consequent hypophosphatemia, phosphaturia and alteration of vitamin D metabolism, leading to the appearance of acquired osteomalacia. Recently a non-phosphaturic variant has been described. This clinical case presents a patient with a non-phosphaturic mesenchymal tumor, whose diagnosis was complicated by nonspecific initial symptoms.
Keywords: mesenchymal tumor, phosphaturia, osteomalacia, FGF-23, diagnosis.
INTRODUCCIÓN
Los tumores mesenquimales fosfatúricos están asociados a osteomalacia oncogénica1. Sin embargo, Folpe y cols en 2004, describieron la variante no fosfatúrica2. La mayoría de tumores son benignos, pequeños y de crecimiento lento. Las localizaciones suelen ser en extremidades y sitios acrales; raramente se presenta como una enfermedad multifocal y las recurrencias tardías ocurren en menos del 5%3. Clínicamente su presentación es variable, pero los síntomas típicos son: dolor óseo, debilidad muscular proximal, pérdida de peso y fracturas múltiples4. Los tumores inductores de osteomalacia (TIO) tienen una producción anormal del factor de crecimiento fibroblástico 23 (FGF-23) que ocasiona la inhibición de la reabsorción de fosfato en el túbulo contorneado proximal y la inactivación de la 1α-hidroxilasa con la consecuente hipofosfatemia, fosfaturia y alteración del metabolismo de la vitamina D que conlleva a la aparición de osteomalacia adquirida5. El diagnóstico y localización de estos tumores suele ser un reto en la práctica clínica, frecuentemente se pasan por alto y son mal diagnosticados en un primer momento. A continuación, se describe el caso de un paciente con un tumor mesenquimal no fosfatúrico cuyo diagnóstico fue difícil dados los síntomas iniciales.
CASO CLÍNICO
Se describe el caso de un paciente masculino de 54 años con antecedentes de diabetes mellitus tipo 2, hipertensión arterial y dislipemia en tratamiento con metformina, imidapril y pitavastatina. En seguimiento por el servicio de Endocrinología por alteración en el metabolismo fosfocálcico en contexto de déficit previo de Vitamina D. Sin antecedentes familiares, ni quirúrgicos de interés. En diciembre de 2014, acude al servicio de Urgencia por dolor y tumefacción en planta de pie derecho sin antecedente traumático, ni episodios previos; realizan radiografía de pie derecho observándose calcificaciones a nivel de sesamoideo, por lo cual se indica tratamiento con AINEs.
Un mes más tarde al notar que el edema y dolor aumentaron de intensidad consulta en el servicio de Traumatología. En la exploración física se encontró dolor selectivo en sesamoideo medial, dorsiflexión dolorosa de primer dedo y presencia de tumoración en región plantar de la cabeza de primer metatarsiano. Fue catalogado como sesamoiditis en estudio y solicitan resonancia magnética, en la cual se observó lesión nodular de partes blandas superficiales a los sesamoideos de 3x2x1 cm en cabeza de primer y segundo metatarsiano con realce periférico heterogéneo (Figura 1), decidiéndose tratamiento sintomático.
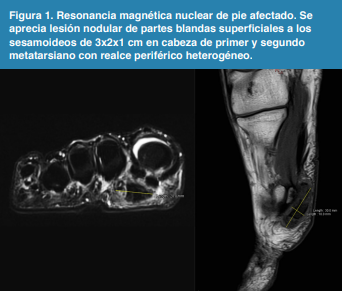
Figura 1
Resonancia magnética nuclear de pie afectado. Se aprecia lesión nodular de partes blandas superficiales a los sesamoideos de 3x2x1 cm en cabeza de primer y segundo metatarsiano con realce periférico heterogéneo.
Tras tres meses de evolución, ante la pobre respuesta al tratamiento analgésico y progresión del dolor con limitación funcional para la deambulación y según los hallazgos de imagen se programa cirugía para exéresis y biopsia de la lesión. En los exámenes preoperatorios solicitados por Endocrinología, se objetiva calcio total 9,3 mg/dl (8,2-10,6 mg/dL), calcio iónico 1,16 mmol/L (0,8-1,2 mmol/L), fósforo 3,4 mg/dl (2,5-5 mg/dl), PTH 40,30 pg/ml (14,5-87,1 pg/ml), sin elevación de fosfaturia, valores normales de 25OH-vitamina D en sangre.
Los resultados de anatomía patológica reportan una masa nodular revestida parcialmente por tejido de aspecto adiposo que mide 2x1,5x0,8 cm. En el estudio histológico la lesión descrita es bien delimitada, no encapsulada, observándose de forma parcheada agregados de células fusiformes con núcleos elongados que se disponen alrededor de segmentos vasculares, con áreas de matriz condroide calcificada. Además, se objetivaron áreas osificadas delimitadas por células gigantes multinucleadas tipo osteoclasto-like, hallazgos compatibles con tumor mesenquimal fosfatúrico variante mixta de tejido conjuntivo.
En la analítica posoperatoria persiste con normofosfatemia, normocalcemia, PTH normal, sin fosfaturia elevada. Actualmente, el paciente puede deambular sin complicaciones y se ha encontrado asintomático.
DISCUSION
Robert McCance en 1947 describió el primer caso de un tumor inductor de osteomalacia. Posteriormente, Prader y cols. en 1957 reportaron la relación entre osteomalacia y tumores de origen mesenquimatoso al descubrir el vínculo entre raquitismo hipofosfatémico y el desarrollo de un tumor costal6.
Más tarde se propuso la existencia de péptidos fosfatúricos elevados en este tipo de tumores al demostrar la inhibición del transporte de fosfato en células epiteliales renales en un medio de cultivo de un tumor obtenido de un paciente con TIO. Entre estos péptidos el FGF-23 es el más descrito, identificándose su expresión en más del 90% de tumores mesenquimales fosfatúricos de variante mixta de tejido conjuntivo y en 75% de los tumores histológicamente idénticos sin TIO. Desde entonces se describe la variante no fosfatúrica como una entidad histopatológica. Bahrami y cols. proponen como posibles explicaciones de la variante no fosfatúrica, la detección temprana de estos tumores, la expresión de niveles bajos de FGF-23, la síntesis de formas no funcionales de FGF-23, ausencia del estudio de los niveles de fosfato antes de la resección y/o la falta de detección de la asociación entre la pérdida ósea y la neoplasia6.
Al ser un diagnóstico poco frecuente suele ser de difícil determinación, en un estudio retrospectivo con 144 pacientes con TIO se encontró que el tiempo que trascurrió entre la aparición de los síntomas y un correcto diagnóstico fue de 2,9±2,3 años. Asimismo, un diagnóstico erróneo inicial sucedió en el 95,1% de los pacientes, siendo los más frecuentes: hernia discal, espondiloartritis y osteoporosis7, en nuestro caso fue sesamoiditis.
La localización de los tumores a menudo es bastante difícil debido a que surgen en huesos o tejidos blandos y suelen ser de pequeño tamaño por lo que se recomienda combinar estudios radiológicos anatómicos y funcionales (con análogos de somatostatina radiomarcados debido a la expresión de dichos receptores)8. Sin embargo, la histopatología es la que permite el diagnóstico definitivo; siendo la variante de tejido conectivo mixta la más común, tal como se muestra en el caso clínico7.
El mejor tratamiento es la resección quirúrgica con márgenes completos lo que lleva a una pronta reversión de las anormalidades bioquímicas y curación de la enfermedad ósea durante un período de 6 a 12 semanas4.
En conclusión, TIO debe ser tomado en cuenta al momento de evaluar a pacientes con síntomas musculoesqueléticos persistentes. Cabe destacar, la importancia de considerar medir niveles de fósforo en la analítica inicial de pacientes con esta sintomatología ya que la hipofosfatemia puede ser la clave para evitar demoras y/o diagnósticos erróneos. Además, si existe la sospecha clínica la medición de FGF-23 puede ser de gran valor para confirmar el diagnóstico, considerando que con la resección quirúrgica completa esta patología es curable.
REFERENCIAS BIBLIOGRÁFICAS
1. Li Y, Li Y, Hui M, Liu Y, Liu X, Jin J, et al. Comparison of surgical treatments of tumor-induced osteomalacia in different locations in the lower limbs: A retrospective study. Medicine (Baltimore). 2019 Mar 1;98(11):e14846.
2. Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY. Most Osteomalacia-associated Mesenchymal Tumors Are a. Am J Surg Pathol. 2004;28(1):1–30.
3. Chong, William, Molinolo, Alfredo, Chen, Clara, Collins M. Tumor-induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
4. Zinan Yin, Juan Du, Fan Yu WX. Tumor-induced osteomalacia. Osteoporos Sarcopenia [Internet]. 2018;4(4):119–27. Available from: https://doi.org/10.1016/j.afos.2018.12.001
5. Halperin F, Anderson RJ, Mulder JE. Tumor-induced osteomalacia: The importance of measuring serum phosphorus levels. Nat Clin Pract Endocrinol Metab. 2007 Oct;3(10):721–5.
6. Bahrami A, Weiss SW, Montgomery E, Horvai AE, Jin L, Inwards CY, et al. RT-PCR Analysis for FGF23 Using Paraffin Sections in the Diagnosis of Phosphaturic Mesenchymal Tumors With and Without Known Tumor Induced Osteomalacia. Am J Surg Pathol. 2009;33(9):1348–54.
7. Feng J, Jiang Y, Wang O, Li M, Xing X, Huo L, et al. The diagnostic dilemma of tumor induced osteomalacia: a retrospective analysis of 144 cases. Endocr J. 2017 Apr 25;64(7):675–83.
8. Florenzano P, Gafni RI, Collins MT. Tumor-induced osteomalacia. Bone Reports. 2017 Jan 1;7:90–7.
Notas de autor
alejandrarivasm@hotmail.com