DEBATE
Monitoramento ambiental na manipulação de medicamentos oncológicos injetáveis à luz das normativas vigentes
Environmental monitoring in compounding injectable oncology drugs as per current legislation
 Priscila da Nobrega Rito
Verônica Viana Vieira
Priscila da Nobrega Rito
Verônica Viana Vieira
Monitoramento ambiental na manipulação de medicamentos oncológicos injetáveis à luz das normativas vigentes
Vigilância Sanitária em Debate, vol. 9, núm. 2, pp. 3-13, 2021
INCQS-FIOCRUZ
Recepção: 18 Outubro 2020
Aprovação: 22 Fevereiro 2021
RESUMO: A manipulação de medicamentos oncológicos injetáveis é uma atividade do segmento farmacêutico de grande relevância e complexidade. Requer ações de biossegurança para minimizar a contaminação ambiental e ocupacional e condições ambientais associadas à técnica asséptica para manutenção da esterilidade. São preparações extemporâneas, isentas de teste de esterilidade, sendo, portanto, necessário um controle rigoroso do processo de preparo. O monitoramento ambiental, exigência da normativa nacional de boas práticas de manipulação, é uma ferramenta utilizada para demonstrar que o ambiente de produção atende às exigências de qualidade. No entanto, a normativa não estabelece a forma de realizá-lo e nem o padrão de conformidade aceitável. Essa ausência de informação propicia a realização de ensaios sem padrão de referência, podendo o produto final ficar aquém das exigências quanto à segurança, integridade e confiabilidade. Nesse sentido, esse trabalho traz à luz as exigências das normativas internacionais quanto ao monitoramento ambiental na manipulação de medicamentos injetáveis traçando um contraponto com as principais normas e guidelines industriais nacionais e internacionais. Embora os produtos industrializados e manipulados por processo asséptico tenham que manter a mesma característica de esterilidade, observam-se divergências de métodos e limites aceitáveis de contaminação, questionando-se se é possível a flexibilização quanto as exigências de qualidade. Ressalta-se ainda a necessidade de a agência regulatória brasileira atualizar a normativa voltada para farmácia de manipulação de injetáveis, para que auxilie na efetiva implantação de um Programa de Monitoramento Ambiental de forma a contribuir para o fortalecimento do Sistema de Gestão da Qualidade em Serviços de Saúde.
Palavras chave: Monitoramento Ambiental, Boas Práticas de Manipulação, Medicamentos Oncológicos Injetáveis.
ABSTRACT: The compounding of injectable oncology drugs is an activity of the pharmaceutical segment of great relevance and complexity. It requires biosafety actions to minimize environmental and occupational contamination, and environmental conditions associated with the aseptic technique for maintaining sterility. They are extemporaneous preparations, exempt from sterility testing, therefore, strict control of the preparation process is necessary. Environmental monitoring in an injectable compounding cleanroom, a requirement of Brazilian legislation that affords good compounding practices, is a tool used to demonstrate that the environment production meets the requirement of quality standards. However, the national regulations do not establish how to do it or the acceptable standard of compliance. This lack of information allows that methods without reference standards exist, and that the final product may be inadequate for the requirements regarding safety, integrity and reliability. As a conclusion, this debate shows the requirements of international regulations regarding environmental monitoring in compounding injectable drugs, drawing a counterpoint with the main national and international industrial standards and guidelines. Although products manufactured and handled by an aseptic process have to maintain the same sterility characteristic, there are divergences in methods and acceptable limits of contamination, questioning whether flexibility is possible in terms of quality requirements. It is also important to highlight the need for the Brazilian regulatory agency to update the rules for the pharmacy for handling injectable drugs, to assist it in the effective implementation of an environmental monitoring program in order to contribute to the strengthening of the Quality Management System in Health Services.
Keywords: Environmental Monitoring, Good Manufacturing Practices, Injectable Oncology Drugs.
INTRODUÇÃO
As doenças e agravos não transmissíveis (DANT) são as principais causas de óbito no mundo. Em 2008, 36 milhões dos óbitos (63%) ocorreram em consequência das DANT, sendo o câncer responsável por 21%. Esses óbitos ocorreram principalmente em países de baixo e médio desenvolvimento, especialmente na população abaixo dos 70 anos1 . A prevenção e o controle dessas doenças em nosso país representam, atualmente, grandes desafios da saúde pública2 .
No Brasil, a estimativa para adultos para o biênio 2020-2022 aponta para a ocorrência de aproximadamente 625 mil casos novos de câncer para cada ano. Os tipos mais frequentes, exceto o câncer de pele não melanoma, serão: próstata, mama, cólon e reto2 .
O câncer infantojuvenil (doença que acomete crianças e adolescentes até 19 anos de idade) é considerado raro quando comparado com os tumores de adulto, correspondendo entre 1% a 4% de todos os tumores malignos. No Brasil, em 2017, foram notificados 2.553 óbitos por neoplasia infantojuvenil, sendo esperados, 8.460 casos novos para cada ano do triênio 2020-20222 .
A terapia anticâncer inclui agentes quimioterápicos, biológicos, terapia alvo molecular, radioterapia, cirurgia e oncologia intervencionista3 . Além da terapia com os medicamentos que agem diretamente no combate à doença, há outras que são empregadas no manejo das toxicidades causadas pelos medicamentos anticâncer: antieméticos, protetores urinários, corticoides e hidratação venosa, incluindo, quando indicado, transfusões de hemácias e plaquetas, antibióticos e fatores de crescimento. O emprego desses fármacos é fundamental para favorecer a continuidade do paciente ao tratamento e evitar a complicação médica e, como consequência, o risco à vida do paciente4 , 5 .
A manipulação de medicamentos injetáveis é uma atividade de grande relevância e de alta complexidade desenvolvida pelo segmento farmacêutico. No âmbito assistencial tem o objetivo de atender uma necessidade do setor de saúde, preparar medicamentos que não são comercialmente disponíveis, a fim de suprir uma necessidade específica do paciente6 , 7 , 8 . É uma importante ferramenta terapêutica que requer conhecimento e experiência do profissional farmacêutico devido à possibilidade de personalização da preparação, como individualização da dose, acréscimo de componentes à formulação, adequação da formulação à via de administração e escolha do tipo e volume de diluente adequado à condição clínica do paciente7 . A manipulação é realizada após a validação farmacêutica da prescrição médica quanto aos componentes da formulação, quanto à dose, à qualidade, à compatibilidade, à estabilidade e às interações com outros medicamentos e/ou alimentos, bem como à viabilidade do tratamento proposto9 . A individualização das formulações na área da oncologia e da nutrição parenteral contribuiu de forma significativa para a evolução da atividade de manipulação pelas farmácias10 , 11 .
As manipulações de injetáveis feitas pelas farmácias são realizadas por técnica asséptica, com o objetivo de prevenir a contaminação das soluções injetáveis, dos insumos e dos correlatos, principalmente por micro-organismos e materiais particulados durante todo o processo de manipulação. No caso de manipulação de medicamentos oncológicos injetáveis, atribuição esta exclusiva do profissional farmacêutico habilitado e indelegável12 , além da técnica asséptica, são fundamentais ações de biossegurança a fim de minimizar o risco ocupacional e a contaminação ambiental10 , 13 , 14 .
Por lei, os medicamentos injetáveis manipulados em farmácias são preparações extemporâneas, ou seja, a infusão da solução deve ser finalizada até 48 h a partir do preparo, sendo dispensadas dos testes de esterilidade e endotoxinas bacterianas13 , logo, o nível de segurança de esterilidade para um produto produzido por processamento asséptico não pode ser previsto como o é para os que passam por esterilização terminal11 .
A esterilidade de um produto produzido por técnica asséptica pode não ser totalmente garantida pelas inúmeras fontes de contaminação que podem ocorrer durante o processo de produção. As fontes podem ser o ar, os funcionários, a água, a infraestrutura da área, o próprio processo, os materiais e os equipamentos utilizados11 , 15 . Para minimizar o risco de danos à saúde do paciente em termos de morbidade e mortalidade, o processo de preparo deve ocorrer em áreas físicas adequadas com sistema de filtragem de ar de alta eficiência (área limpa), onde o número de partículas viáveis e não viáveis presentes é conhecido e controlado, sendo fundamental que todos os processos e condutas assépticas sejam validados e seguidos rigorosamente pelos funcionários envolvidos na produção e que haja a contínua capacitação desses e atualização dos processos11 , 16 .
O monitoramento ambiental (partículas viáveis e não viáveis) em uma área limpa de produção de injetáveis é uma ferramenta do programa de garantia da qualidade utilizada para demonstrar que o ambiente de produção de injetáveis atende às exigências de qualidade para o objetivo proposto. Deve ser empregado como um indicador de qualidade dos equipamentos, da infraestrutura da área e dos processos. Permite verificar a eficiência do sistema de filtragem de ar, acompanhar as práticas assépticas dos funcionários envolvidos na produção e avaliar o desempenho dos processos de limpeza e desinfecção. Portanto, o monitoramento torna-se absolutamente necessário em processos assépticos a fim de demonstrar o controle da microbiota do ambiente de preparo de injetáveis, e por permitir ações preventivas antes da contaminação ocorrer11 , 17 , 18 .
A normativa brasileira para boas práticas de manipulação de medicamentos injetáveis13 faz exigência de programa de monitoramento ambiental. No entanto, não estabelece a forma de realizá-lo e nem o padrão de conformidade aceitável. Essa lacuna de informação propicia a realização de ensaios para o monitoramento ambiental microbiológico sem padrão de referência. Sem este, o produto final fica aquém das exigências quanto à segurança, integridade, e confiabilidade do produto19 , 20 .
Visto isso, há a necessidade de busca dessas informações em normativas e guidelines voltados à farmácia de manipulação internacional e/ou à indústria farmacêutica. Nesse sentido, esse trabalho traz à luz as exigências das normativas internacionais quanto ao monitoramento ambiental na manipulação de medicamentos injetáveis em farmácias traçando um contraponto com as principais normas e guidelines industriais nacionais e internacionais.
No entanto, a utilização de parâmetros de monitoramento ambiental de diretrizes industriais, em que é exigido alto rigor no processo de preparo de medicamentos e fiscalização pós-produção a fim de garantir a qualidade total do produto é aplicável ao processo de manipulação de medicamento injetável, cujo objetivo é atender um paciente vulnerável quanto a sua condição de imunossupressão decorrente do tratamento oncológico, ou é possível uma flexibilização de métodos e parâmetros de conformidade?
Exigências das normativas nacionais quanto às boas práticas de manipulação de medicamentos oncológicos injetáveis em farmácias
Em 21 de setembro de 2004, foi publicada pela Agência Nacional de Vigilância Sanitária (Anvisa) a Resolução da Diretoria Colegiada (RDC) nº 22021 , primeira normativa que estabeleceu os requisitos necessários de funcionamento dos serviços que oferecem terapia antineoplásica tanto em serviço público quanto privado. A Normativa estabeleceu as orientações gerais para as Boas Práticas de Preparação e Administração da Terapia Antineoplásica (BPPTA), determinou a formação da equipe multidisciplinar, a obrigatoriedade de um Programa de Controle Médico de Saúde Ocupacional e os cuidados de biossegurança a fim de preservar a saúde ocupacional e ambiental21 .
A RDC nº 67, de 8 de outubro de 200713 e suas atualizações (RDC nº 87, de 21 de novembro de 2008, e RDC nº 21, de 20 de maio de 2009) dispõem sobre as Boas Práticas de Manipulação de Preparações Magistrais e Oficinais para Uso Humano em farmácias tanto no serviço público quanto no privado. Essa Resolução, que está em vigor, abrange não só a manipulação de medicamentos estéreis e não estéreis oncológicos, mas também a manipulação de medicamentos a partir de insumos/matérias primas, inclusive de origem vegetal, de substâncias de baixo índice terapêutico, de antibióticos, hormônios e substâncias sujeitas à controle especial e de medicamentos homeopáticos, assim como, a manipulação de doses unitárias e unitarização de dose de medicamentos em serviços de saúde. Exclui da sua regulamentação a manipulação de nutrição parenteral, enteral e concentrado polieletrolítico para hemodiálise.
A RDC nº 67/2007 revoga as RDC nº 214, de 12 de dezembro de 2006, e nº 354, de 18 de dezembro de 2003, a fim de padronizar e uniformizar por igual todo o setor magistral brasileiro19 . Com o intuito de garantir a qualidade do produto final e a segurança ao paciente, requisitos fundamentais para restabelecer a credibilidade do setor magistral foram ampliados. Um dos requisitos se refere às condições mínimas para o preparo dos medicamentos que atingem questões relacionadas a todas as etapas de produção como: infraestrutura, instalações e equipamentos adequados, recursos humanos suficientes e capacitados, controle de qualidade nas etapas do processo produtivo, avaliação farmacêutica da prescrição, manipulação, conservação, armazenamento, transporte, dispensação das preparações e atenção farmacêutica visando a garantia da qualidade, segurança e eficácia do produto de forma a promover o uso seguro desses medicamentos à população22 .
Embora as duas normativas citadas abordem as boas práticas de manipulação de medicamentos oncológicos estéreis, observa-se um maior rigor da RDC nº 67/2007 principalmente quanto aos critérios de infraestrutura e instalação, como a exigência de uma área limpa com seus controles de partículas, temperatura e umidade, presença de antecâmaras para minimizar a possibilidade de contaminação entre ambientes, exigência de materiais de construção e de mobiliários adequados a fim de evitar acúmulo de partículas viáveis e não viáveis e facilitar a limpeza e desinfecção13 , 17 . Quanto aos critérios de qualidade, exige validação de processos a fim de garantir a reprodutibilidade dos procedimentos por todos os manipuladores e um programa de monitoramento ambiental a fim de avaliar o nível de contaminação do ar e superfícies do ambiente de manipulação de medicamentos estéreis13 . Na Tabela 1 estão descritas algumas dessas exigências.
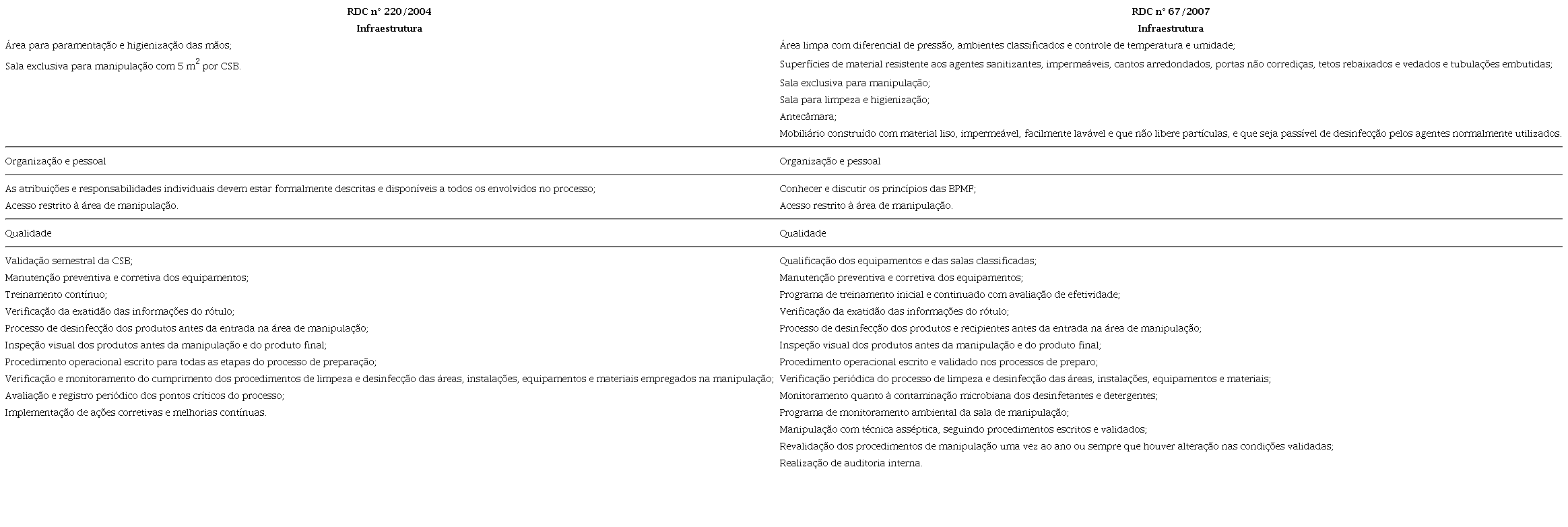
BPMF: Boas Práticas de Manipulação em Farmácias; CSB: Cabine de Segurança Biológica.
Qualidade na preparação de medicamentos injetáveis e o controle sanitário
O gerenciamento da qualidade é um conceito abrangente, que cobre todas as questões que determinam, isolada ou conjuntamente, a qualidade de um produto. Corresponde à soma dos arranjos organizados com o objetivo de garantir que os medicamentos tenham a qualidade exigida para o uso pretendido23 .
A qualidade aborda questões relacionadas à qualificação de sistemas, instalação e equipamentos, validação de processo, controle de qualidade laboratorial, certificação de qualidade dos materiais empregados, treinamento dos funcionários, monitoramento ambiental periódico, investigação de desvios e agilidade nas ações corretivas e preventivas (CAPA)23 . Esses elementos fazem parte da garantia da qualidade, inserida na gestão da qualidade, uma ferramenta proativa em processos assépticos, presente na indústria farmacêutica e com papel fundamental durante todas as etapas de produção a fim de garantir a esterilidade do produto final17 . A garantia da qualidade exige conhecimento, equipe multidisciplinar, equipamentos, investimento e treinamento, o que pode dificultar sua execução pelas farmácias que manipulam medicamentos injetáveis22 , 24 .
As exigências cada vez maiores a respeito da qualidade no preparo dos medicamentos manipulados pelas autoridades sanitárias geram polêmicas quanto ao custo necessário para dispor e adequar a área física para manipulação. As exigências de realizar todos os processos de controles de qualidades durante as etapas de produção, treinamento dos funcionários e a limitação de recursos financeiros quando comparado aos da indústria farmacêutica criam a sensação da oferta à população de um medicamento com menor rigor de qualidade. No entanto, o binômio de qualidade, segurança e eficácia, é indissociável, não devendo ser tratado como um requisito legal, e sim como essencial, um atributo inerente ao produto, construído durante os processos. Esta é a forma mais efetiva de garantir a segurança do usuário e a efetividade do produto17 , 25 , 26 , 27 .
Embora o medicamento injetável manipulado em farmácias seja preparado conforme recomendação do fabricante, em que as estabilidades físico-química e microbiológica do produto final são definidas pelo fabricante, a garantia da estabilidade microbiológica após a abertura do frasco ou ampola de medicamento é dependente das condições do ambiente de manipulação e do rigor das condutas empregadas pelos trabalhadores durante os processos. Diante disso, a fase de validação de processos, treinamento dos funcionários e implementação de um programa de monitoramento ambiental, requisitos de qualidade exigidos pela RDC nº 67/2007, é um desafio fundamental à garantia da qualidade e exigido ao atendimento as boas práticas de manipulação de medicamentos. No entanto, a exigência por lei por si só não garante segurança na manipulação22 . A lei tem o intuito de tornar a atividade mais profissional, mais científica e segura, e o profissional deve entender essas exigências como fomento à qualidade dos processos. Por sua vez, é fundamental um controle sanitário com fiscalização para obrigar, com rigor, que os estabelecimentos atendam aos requisitos mínimos exigidos pela legislação, mas de forma não apenas punitiva, mas parceira e participativa a fim de contribuir para construção de práticas mais seguras do cuidado assistencial de saúde22 , 28 , 29 .
Monitoramento ambiental em áreas limpas de produção de medicamentos injetáveis
O monitoramento ambiental em uma área limpa é uma ferramenta do programa de garantia da qualidade e, quando planejado e executado de forma rigorosa, permite aumentar o nível de qualidade do ambiente de produção de injetáveis, principalmente os de processamento asséptico16 , 23 , 30 . O objetivo é identificar a estabilidade e as mudanças no cenário da microbiota do ambiente de produção a fim de demonstrar alterações ou falhas nos processos11 , 31 .
Devido à insuficiência de informações nas normativas brasileiras vigentes (RDC n° 67/2007 e RDC n° 220/2004) a respeito dos ensaios necessários para o monitoramento ambiental em áreas limpas de farmácias que manipulam injetáveis, bem como os limites de contaminação e condutas em caso de desvio, faz-se necessária a consulta nas diretrizes internacionais que orientem quanto ao monitoramento em áreas limpas de manipulação, como a Farmacopeia Americana14 , 32 , o Guia Europeu desenvolvido pela Pharmaceutical Inspection Co-operation Scheme (PIC/S)33 e normativas e diretrizes internacionais e nacionais voltadas para a fabricação de medicamentos injetáveis, como as da União Europeia34 , dos Estados Unidos31 , da Organização Mundial da Saúde (OMS)30 e do Brasil18 , 23 , 35 , a fim de atender às premissas de qualidade e segurança ambiental.
O monitoramento ambiental é a realização de ensaios em operação e em repouso. O ensaio em operação é considerado o mais importante por avaliar as práticas assépticas realizadas pelos funcionários e por demonstrar se a classificação da área para um determinado processo, com os equipamentos ligados e fluxo de materiais e funcionários, é mantida durante as atividades de produção34 , 35 , 36 . O ensaio em repouso deve ser realizado ao término das atividades e sem os funcionários presentes. Permite verificar se o sistema de filtragem de ar da área limpa é capaz de se restabelecer em um curto período de tempo e conhecer a base da microbiota do ambiente após a desocupação da área e a realização dos procedimentos de limpeza e desinfecção35 , 36 , 37 , 38 .
Visa acompanhar a qualidade do ambiente de forma contínua principalmente durante as fases críticas do processo produtivo, permitindo identificar os momentos de risco de contaminação, possibilitando ações para prevenção da contaminação do produto. Os resultados do monitoramento permitem criar ferramentas como gráficos de tendência histórica e estabelecer limites de alerta e ação para os parâmetros que se quer acompanhar. Logo, o monitoramento avalia não só o desempenho do sistema de filtragem de ar da área de manipulação, mas também a estrutura física da área, procedimentos de paramentação e limpeza dos funcionários, funcionamento dos equipamentos e seguimento criterioso dos processos18 , 35 , 36 , 39 .
O monitoramento ambiental engloba o monitoramento físico (partículas não viáveis, diferencial de pressão, temperatura e umidade) e microbiológico (partículas viáveis)18 , 33 , 40 .
O monitoramento de partículas não viáveis é realizado através da contagem de partículas totais de diâmetro acima de 0,5 µm e 5,0 µm suspensas no ar. Os micro-organismos são carreados pelo ar associados, geralmente, a partículas de diâmetro entre 10,0-20,0 µm. Portanto, esse tipo de monitoramento não tem o objetivo de avaliar o conteúdo microbiológico18 , mas sim a qualidade e a possibilidade de contaminação do ambiente de produção.
O diferencial de pressão, direcionamento do fluxo de ar entre diferentes ambientes em uma área limpa, é um parâmetro importante a ser avaliado durante o processo de produção31 , 40 , a fim de evitar a transferência de ar contaminado entre ambientes18 , 41 . Para o preparo de medicamentos oncológicos injetáveis é exigido um ambiente com pressão negativa13 . Nesse caso, o diferencial de pressão da área de produção deve ser inferior ao das áreas adjacentes, garantindo que possíveis contaminantes químicos e tóxicos formados durante o processo de produção não sejam capazes de contaminar as áreas adjacentes e ao mesmo tempo impedir que contaminantes das áreas adjacentes, principalmente microbiológico, alcancem à área de produção14 , 18 , 41 .
Outros parâmetros como temperatura e umidade, cruciais por inibir a proliferação microbiológica no ambiente de produção e garantir ambiente confortável aos funcionários, devem ser monitorados regularmente. A temperatura de conforto deve considerar o tipo de roupa utilizada para as atividades realizadas na área limpa a fim de minimizar a liberação de partículas pelos funcionários30 , 35 . Os ensaios, parâmetros e métodos para monitoramento físico são realizados conforme estabelecido pela International Organization for Standardization (ISO)42 .
O monitoramento de partículas viáveis é realizado com o objetivo de controlar o conteúdo microbiológico na área de produção, dentro de limites específicos, através da amostragem ativa e passiva do ar, das superfícies e dos funcionários11 , 18 . A avaliação qualitativa (caracterização e a identificação) da microbiota presente na área de produção permite conhecer as características dos micro-organismos quanto à capacidade de resistência e patogenicidade, identificar as possíveis fontes de contaminação e correlacionar com práticas de limpeza, desinfecção e higiene dos funcionários, permitindo o planejamento de ações efetivas32 , 43 .
A definição da frequência, o número de pontos a serem amostrados e os locais críticos para o processo de produção devem ser baseados, preferencialmente, em uma análise de risco18 , 31 . A análise de risco em uma determinada área de processamento asséptico torna o programa de monitoramento ambiental mais significativo e permite orientar a amostragem para locais do processo em que o risco de contaminação é maior. A partir dessa análise, é possível estabelecer a frequência de amostragem baseada nos níveis de risco para cada área ou processo44 . No caso da impossibilidade de realizar, os locais a serem mapeados devem representar toda a área e considerar principalmente os pontos próximos à área crítica (Grau A), o posicionamento, circulação, vestimenta e mãos dos funcionários, já que são considerados os principais carreadores de contaminação dentro do processo produtivo. Os locais de entrada e saída de materiais e equipamentos de uma área de menor classificação, quanto à partícula total, para uma área de maior classificação, também devem estar incluídos no plano de amostragem32 .
Exigências para execução de um programa de monitoramento ambiental em áreas limpas de produção de medicamentos em indústrias e de manipulação de injetáveis em farmácias
A definição de frequência de amostragem para partículas viáveis deve ser determinada a partir de uma análise de risco, no entanto diretrizes para indústria farmacêutica18 , 31 , 34 sugerem o monitoramento rigoroso em todos os turnos de produção em ambientes considerados críticos como em Grau A e, à medida que o rigor do ambiente diminui (Grau C e D), a frequência de amostragem também pode reduzir18 . As diretrizes internacionais voltadas para farmácias que manipulam injetáveis32 , 33 sugerem uma frequência mínima de amostragem, conforme apresentado na Tabela 2 .

UFC: unidades formadoras de colônias; ND: valor não definido.
Foram verificadas divergências quanto à frequência sugerida para amostragem entre as diretrizes para farmácia ( Tabela 2 ) e limites de contaminação tanto nas diretrizes voltadas para indústria quanto para farmácias ( Tabela 3 ).

EU GMP: European Union Guidelines to Good Manufacturing Practice ; Anvisa: Agência Nacional de Vigilância Sanitária; WHO: World Health Organization ; FDA GMP: Food and Drug Administration Guidance for Industry – Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice ; UFC: unidades formadoras de colônias: ND: valor não definido. *Amostras de áreas Grau A deveriam normalmente não ter nenhum contaminante microbiológico.
A Farmacopeia Americana sugere uma maior frequência para o monitoramento em ambientes críticos e adjacentes (Grau A e C), seguindo a mesma orientação das diretrizes industriais, quando comparada ao Guia Europeu (PIC/S)33 para farmácias com manipulação de injetáveis. No entanto, este mesmo Guia33 exige o mesmo limite rigoroso de contaminação microbiológica que a diretriz europeia ( EU Guidelines to Good Manufacturing Practice: Medicinal Products for Human and Veterinary Use ) para monitoramento ambiental em industrias34 . Se em um ambiente é exigido rigor quanto ao crescimento de microrganismo, assim como o é para indústria farmacêutica, esse ambiente deveria ser monitorado com mesmo padrão para se afirmar que o processo de manipulação está em conformidade e que o risco de contaminação é reduzido. Logo, pode ser considerado um contrassenso sugerir uma frequência menor de monitoramento em ambientes em que o crescimento microbiológico pode levar danos ao paciente.
Para o ensaio de amostragem ativa de ar, em um ambiente Grau C, as normativas americanas ( Food and Drug Administration - Good Manufacturing Practice 31 e Farmacopeia Americana14 ) estabelecem o limite de ação a partir de 10 unidades formadoras de colônias (UFC), e as demais normativas18 , 30 , 33 , 34 a partir de 100 UFC. Em relação às amostragens de superfícies e das luvas do manipulador, a normativa americana para indústria31 não especifica limite de contaminação, já a Farmacopeia Americana14 permite, em ambiente Grau A, até 3 UFC/placa e as outras diretrizes nacionais e internacionais18 , 30 , 33 , 34 não permitem nenhum crescimento microbiano. Portanto, foi observada uma contradição dos limites estabelecidos pela Farmacopeia Americana quando comparada às outras diretrizes, por permitir um limite maior de contaminação, em ambientes considerados críticos ao processo produtivo (Grau A). Considerando que a manipulação de injetáveis ocorre por processo asséptico em um ambiente Grau A, que são preparações extemporâneas e não passam pela etapa de esterilização terminal e nem por testes de esterilidade, seria imprudente permitir um maior limite de contaminação nesses ambientes considerados críticos.
Quanto à amostragem passiva do ar, o guia18 e a Instrução Normativa35 , ambos da Anvisa, exigem esse ensaio em todos os ambientes de produção, variando apenas a frequência de amostragem de acordo com a classificação da área. Em ambientes mais críticos (Grau A e B), a amostragem deve ser feita durante todo o processo produtivo, já em ambientes com baixo risco de contaminação para o produto, a amostragem deve ser semanal ou até mensal (Grau C e D). As diretrizes americanas para farmácia industrial31 e para farmácia com manipulação14 , 32 não exigem o uso desse método de amostragem. Descrevem o ensaio como opcional, porém, devido ao resultado semiquantitativo ou qualitativo desse método, não deve ser empregado de forma isolada, e sim associado a outros métodos de amostragem. As divergências de opiniões quanto ao emprego desse método também se estendem em vários artigos publicados45 , 46 , 47 . Portanto, mesmo com as limitações do método, esse ensaio deve ser considerado na elaboração de um plano de monitoramento para se adequar às exigências da normativa brasileira16 , 18 , 35 . Porém, considera-se importante um estudo amplo para a avaliação do método de amostragem passiva a fim de fundamentar a real obrigatoriedade de tal ensaio pela legislação nacional.
O ensaio para a amostragem das luvas dos funcionários em ambiente Grau C está apenas descrito no Guia Europeu33 para farmácias que manipulam medicamentos injetáveis. No entanto, este guia não estabelece o limite de contaminação das luvas nesse ambiente, ficando o ensaio sem parâmetro. Embora esse ensaio não esteja descrito nos guias de monitoramento para a indústria farmacêutica dos Estados Unidos31 , da Europa34 e da OMS30 , documento técnico de associação internacional48 , na Farmacopeia Brasileira16 , Americana14 , 32 e Japonesa49 e nas normativas nacionais18 , 23 , 35 , deve-se avaliar a importância da realização deste ensaio por farmácias que manipulam medicamentos injetáveis, uma vez que a área Grau C é o ambiente mais próximo à área Grau A, conforme exigência da RDC nº 67/2007. O funcionário que trabalha neste ambiente (Grau C) está diretamente em contato com os materiais (estéreis e não estéreis) que são introduzidos na Cabine de Segurança Biológica (Grau A). Desta forma, o risco de carreamento de contaminantes provenientes desses materiais para o interior do ambiente Grau A, por meio das mãos dos funcionários, é alto, principalmente quando práticas assépticas e de limpeza e desinfecção desses materiais são mal conduzidas50 . A partir disso, foi realizada uma busca nas edições da Farmacopeia Americana e visto que até 2011, o capítulo <1116> Microbiological Control and Monitoring of Aseptic Processing Environments aceitava um limite máximo de crescimento de micro-organismos nas luvas dos funcionários em área Grau C de até 10 UFC/placa51 . Em 2012, o capítulo <1116> foi completamente revisado e com a atualização foi proposta uma análise diferente para a tomada de ações em casos de desvios. Essa nova proposta considera a taxa de incidência da contaminação durante um determinado período e exclui os limites aceitáveis em valores absolutos para crescimento de micro-organismo nas amostragens realizadas no ar, superfícies e funcionários. A justificativa para essa mudança radical na forma de avaliar os resultados de contaminação se deve a limitação dos ensaios empregados na recuperação da microbiota presente no ambiente de produção e a baixa significância em valores absolutos de UFC entre o limite de um resultado conforme e não conforme. Todavia, considerando os riscos de carreamento de contaminantes por meio das mãos dos funcionários50 , podemos considerar fundamental que esse tipo de ensaio seja definido pela normativa nacional, assim como o limite aceitável de contaminação a fim de ser um indicador de condutas de higiene e de processo.
Com relação aos ensaios físicos, os guias e as normas industriais da Europa34 , da OMS30 , do Japão40 e da Anvisa18 , 35 são unânimes ao exigir monitoramento contínuo de partículas não viáveis em ambientes Grau A durante todo o processo de produção. Esse tipo de monitoramento é capaz de mostrar a quantidade de partículas totais no momento da execução da produção. Logo, é um indicador em tempo real de qualidade do ambiente em que os processos estão sendo realizados, permitindo que as não conformidades sejam corrigidas a tempo de não contaminar o produto. No entanto, as diretrizes voltadas para farmácias divergem quanto a este monitoramento. A Farmacopeia Americana14 exige monitoramento semestral e o Guia Europeu33 , monitoramento trimestral ( Tabela 4 ). A frequência de monitoramento sugerida por essas duas diretrizes avalia o desempenho do sistema de filtragem de ar e dos processos realizados no momento específico de realização do ensaio, ficando o período sem ensaio descoberto quanto à informação sobre a qualidade do ambiente de produção, não sendo possível identificar não conformidades e de impedir que um produto contaminado possa alcançar o paciente.

A ISO 14.64452 é utilizada como referência para classificação de áreas limpas para as indústrias de biotecnologia, alimentos, microeletrônica e espacial, não sendo específica para indústrias farmacêuticas. Isso permite que áreas limpas com diferentes finalidades sigam a mesma especificação técnica e de desempenho e tenham os mesmos critérios de avaliação11 . Porém, a ISO 14.64452 não estabelece os níveis de carga microbiana (partículas viáveis), critério importante para a indústria farmacêutica. Além disso, não define os limites para as partículas em suspensão no ar quanto aos estados ocupacionais “em repouso” e “em operação”18 , 53 , por isso, os guias industriais para fabricação de medicamentos continuam a ser utilizados como referência para a produção asséptica em área limpa11 . Os limites de partículas em suspensão para cada classificação de ambiente em repouso e em operação e o diferencial de pressão entre ambientes estão descritos na Tabela 5 .
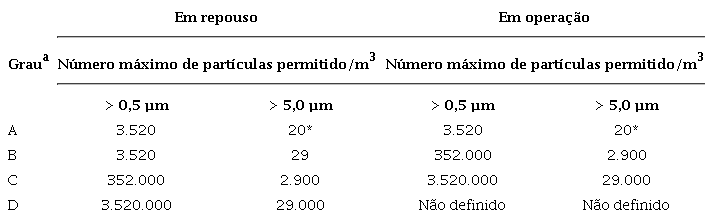
*A partir de 2015, a ISO 14.644-1 não define o valor máximo para partículas acima de 5 mm em ambiente Grau A. a O diferencial de pressão entre dois ambientes pode variar de 5-20 Pa.
O Guia Europeu (PIC/S)33 para farmácias exige os mesmos resultados de partículas totais que a diretriz europeia para indústria farmacêutica34 ( EU Guidelines to Good Manufacturing Practice: Medicinal Products for Human and Veterinary Use), tanto em repouso quanto em operação, diferentemente da Farmacopeia Americana14 e da RDC nº 67/200713 , que exigem apenas esse ensaio físico a cada seis meses e em repouso, ou seja, ensaios para avaliar o desempenho do sistema, ensaios de requalificação.
O monitoramento de partículas totais, diferentemente do de partículas viáveis, fornece resultado imediato, não sendo necessário incubar e esperar de 2 a 5 dias para verificar se um ambiente está de acordo ou não com a especificação para, então, atuar de forma a identificar, corrigir o problema e prevenir a sua recorrência. Logo, seria prudente intensificar esse tipo de monitoramento físico a fim de se conhecer a verdadeira situação do ambiente em que o produto está sendo manipulado.
Na elaboração desse debate, foi observado que não há um documento único que contemple todas as informações necessárias para o monitoramento ambiental em uma área de produção de medicamentos injetáveis. No entanto, as informações para o monitoramento ambiental em produção industrial são mais consolidadas e uniformes entre os documentos técnicos, diferentemente do que ocorre nos documentos para monitoramento ambiental em farmácias que manipulam medicamentos injetáveis. Essas divergências não se restringem somente às diretrizes para farmácias, mas ocorrem também entre as diretrizes da indústria e as da farmácia. Essas divergências são injustificáveis, pois a manipulação de medicamentos injetáveis em farmácias ocorre por técnica asséptica, depende do desempenho do sistema de filtragem da área limpa, das condutas rigorosas exercidas pelos funcionários, da validação de processos e da calibração de equipamentos, assim como para a indústria.
Assim, pode-se afirmar que não há justificativa para flexibilização das exigências de qualidade em relação aos medicamentos manipulados, cabendo à Política Nacional de Vigilância Sanitária de medicamentos assegurar uma oferta terapêutica nacional de qualidade, respeitando os atributos de segurança e eficácia dos medicamentos disponíveis no país26 .
CONCLUSÕES
Diante do exposto, verifica-se a ausência de um documento que contenha todos os parâmetros necessários para o monitoramento ambiental em uma área de manipulação de medicamentos injetáveis. Dessa forma, ressalta-se a necessidade de a agência regulatória brasileira se atentar para a questão da atualização da normativa voltada para farmácias que manipulam medicamentos injetáveis, para que diretrizes de orientação aos profissionais quanto à elaboração de um programa de monitoramento ambiental e a sua efetiva implantação sejam estabelecidas.
Portanto, considera-se fundamental a criação de padrões de referência para orientar profissionais da área a fim de que sejam estabelecidos procedimentos e condutas reprodutíveis e controladas de forma a contribuir para o fortalecimento do Sistema de Gestão da Qualidade nos Serviços de Saúde.
REFERÊNCIAS
1. World Health Organization – WHO. Global action plan for the prevention and control of noncommunicable diseases 2013-2020. Geneva: World Health Organization; 2013[acesso 3 ago 2020]. Disponível em: http://apps.who.int/iris/bitstream/handle/10665/94384/9789241506236_eng.pdf;jsessionid=50BB17F7DAB2DCE4ABD644FC6A16DFEF?sequence=1
2. Instituto Nacional de Câncer José Alencar Gomes da Silva – INCA. Estimativa 2020: incidência de câncer no Brasil. Rio de Janeiro: Instituto Nacional de Câncer José Alencar Gomes da Silva; 2019.
3. Sag AA, Selcukbiricik AF, Mandel NM. Evidence-based medical oncology and interventional radiology paradigms for liver-dominant colorectal câncer metástases. World J Gastroenterol. 2016;22(11):3127-49. http://doi.org/10.3748/wjg.v22.i11.3127
4. Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 2018;47(19):6645-53. http://doi.org/10.1039/c8dt00838h
5. Matz EL, Hsieh MH. Review of advances in uroprotective agents for cyclophosphamide and ifosfamide-induced hemorrhagic cystitis. Urology. 2017;100:16-9. http://doi.org/10.1016/j.urology.2016.07.030
6. American Society of Health-System Pharmacists – ASHP. ASHP guidelines on compounding sterile preparations. Am J Health Syst Pharm. 2014;71(2):145-66. http://doi.org/10.2146/sp140001
7. Mohiuddin AK. Extemporaneous compounding: cautions, controversies and convenience. IP Int J Compr Adv Pharmacol. 2018;3(4):124-37. http://doi.org/10.18231/2456-9542.2018.0028
8. Pergolizzi JV, Labhsetwar S, Lequang JA. Compounding pharmacies: who is in charge? Pain Pract. 2013;13(3):253-7. http://doi.org/10.1111/papr.12033
9. Conselho Federal de Farmácia – CFF. Resolução Nº 565, de 6 de dezembro de 2012. Dispõe sobre a competência legal para exercício da manipulação de drogas antineoplásicas pelo farmacêutico. Diário Oficial União. 7 dez 2012.
10. Myers CE. History of sterile compounding in US hospitals: learning from the tragic lessons of the past. Am J Health Syst Pharm. 2013;70(16):1415-27. http://doi.org/10.2146/ajhp130112
11. Pinto TSA, Kaneko TM, Pinto AF. Controle de produtos estéreis: ênfase nos processos assépticos. In: Pinto TSA, Kaneko TM, Pinto AF. Controle biológico de qualidade de produtos farmacêuticos, correlatos e cosméticos. 4a ed. São Paulo: Manole; 2015. p. 387-448.
12. Conselho Federal de Farmácia – CFF. Resolução Nº 640, de 27 de abril de 2017. Dá nova redação ao artigo 1 da resolução Nº 623 de 2016, estabelecendo titulação mínima para a atuação do farmacêutico em oncologia. Diário Oficial União. 8 maio 2017.
13. Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC Nº 67, de 8 de outubro de 2007. Dispõe sobre boas práticas de manipulação de preparações magistrais e oficinais para uso humano em farmácias. Diário Oficial União. 9 out 2007.
14. The United States Pharmacopeia – USP. Pharmaceutical compounding: sterile preparations. 40a ed. Rockville: The United States Pharmacopeial; 2017.
15. Gapp G, Holzknecht P. Risk analyses of sterile production plants: a new and simple, workable approach. PDA J Pharm Sci Technol. 2011;65(3):217-26. http://doi.org/10.5731/pdajpst.2011.00693
16. Agência Nacional de Vigilância Sanitária – Anvisa. Farmacopeia brasileira. In: Agência Nacional de Vigilância Sanitária – Anvisa. Preparação de produtos estéreis. Brasília: Agência Nacional de Vigilância Sanitária; 2010. 321-37.
17. Shintani H. Validation study on how to avoid microbial contamination during pharmaceutical production. Biocontrol Science. 2015;20(1):1-10. http://doi.org/10.4265/bio.20.1
18. Agência Nacional de Vigilância Sanitária – Anvisa. Guia de qualidade para sistemas de tratamento de ar e monitoramento ambiental na indústria farmacêutica. Brasília: Agência Nacional de Vigilância Sanitária; 2013.
19. Almeida MLC, Nascimento Filho AP. Análise e discussão de aspectos críticos da resolução 67/2007 da Anvisa para as farmácias com manipulação. Infarma. 2010;22(11/22):13-24.
20. Agência Nacional de Vigilância Sanitária – Anvisa. Informes técnicos institucionais: subsídios à discussão sobre a proposta de regulamentação para farmácias magistrais. Rev Saúde Pública. 2005;39(4):691-4. http://doi.org/10.1590/S0034-89102005000400028
21. Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC Nº 220, de 21 de setembro de 2004. Aprova o regulamento técnico de funcionamento dos serviços de terapia antineoplásica. Diário Oficial União. 23 set 2004.
22. Bonfilio R, Emerick GL, Netto Júnior A, Salgado HRN. Farmácia magistral: sua importância e seu perfil de qualidade. Rev Baiana Saúde Pública. 2010;34(3):653-64. http://doi.org/10.22278/2318-2660.2010.v34.n3.a63
23. Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC Nº 301, de 21 de agosto de 2019. Dispõe sobre as diretrizes gerais de boas práticas de fabricação de medicamentos. Diário Oficial União. 22 abr 2020
24. Rodrigues RHRM. Avaliação do controle de qualidade realizado nas farmácias de manipulação de medicamentos e as ações de vigilância sanitária no município de Campo Grande, Mato Grosso do Sul [dissertação]. Rio de Janeiro: Fundação Oswaldo Cruz; 2010.
25. Silva RF, Nascimento Filho AP, Mendonça DC. Estratégias competitivas no mercado farmacêutico brasileiro: uma abordagem sobre o setor magistral. In: Anais do 13º Simpósio de Engenharia de Produção; Bauru, Brasil. Bauru: Universidade Estadual Paulista; 2006.
26. Silva ACP, Oliveira CVS, Cavalheiro MVS, Miranda MCC. Desafios para a rede nacional de laboratórios de vigilância sanitária: o caso dos medicamentos manipulados. Cienc Saúde Coletiva. 2010;15(Supl.3):3371-80. http://doi.org/10.1590/S1413-81232010000900012
27. Mota VLM, Oshiro Junior JA, Chiari-Andréo BG. O controle da contaminação microbiológica de produtos magistrais. Rev Bras Multidiscip. 2017;20(1):33-47. http://doi.org/10.25061/2527-2675/ReBraM/2017.v20i1.474
28. Packer JF, Frota OP. Vigilância sanitária aplicada ao setor magistral [especialização]. Campo Grande: Universidade Católica Dom Bosco; 2016[acesso 17 jul 2020]. Disponível em: http://www.packerconsultoria.com.br/wp-content/uploads/2016/10/janaina_fernanda_packer_banca.pdf
29. Auxiliadora E, Costa M. Vigilância sanitária em serviços de saúde: os desafios da prática. Vigil Sanit Debate. 2014;2(2):27-33. http://doi.org/10.3395/vd.v2i2.148
30. World Health Organization – WHO. Good manufacturing practices for sterile pharmaceutical products. Geneva: World Health Organization; 2011.
31. US Food and Drugs Administration – FDA. Guidance for industry: sterile drug products produced by aseptic processing: current good manufacturing practice. Rockville: US Food and Drugs Administration; 2004.
32. The United States Pharmacopeia – USP. Microbiological control and monitoring of aseptic processing environments. 40a ed. Rockville: The United States Pharmacopeial; 2017.
33. Pharmaceutical Inspection Convention – PIC. PIC/S guide PE 010: guide to good practices for the preparation of medicinal products in healthcare establishments. Geneva: Pharmaceutical Inspection Convention; 2014[acesso 5 jul 2020]. Disponível em: http://www.picscheme.org
34. European Comission – EU. EU guidelines to good manufacturing practice: medicinal products for human and veterinary use. Brussell: European Comission; 2004.
35. Agência Nacional de Vigilância Sanitária – Anvisa. Instrução normativa Nº 35, de 21 de agosto de 2019. Dispõe sobre as boas práticas de fabricação complementares a medicamentos estéreis. Diário Oficial União. 22 ago 2020.
36. White W. Cleanroom technology: fundamental of design, testing and operation. London: Wiley; 2003.
37. World Health Organization – WHO. Environmental monitoring of clean rooms in vaccine manufacturing facilities: points to consider for manufacturers of human vaccines. Geneva: World Health Organization; 2012[acesso 10 jul 2020]. Disponível em: http://www.who.int/immunization_standards/vaccine_quality/env_monitoring_cleanrooms_final.pdf
38. Sandle T. Application of quality risk management to set viable environmental monitoring frequencies in biotechnology processing and support areas. PDA J Sci and Tech. 2012;66(6):560-79. http://doi.org/10.5731/pdajpst.2012.00891
39. International Organization for Standardization – ISO. ISO 14644 cleanrooms and associated controlled environments: part 2: monitoring to provide evidence of cleanroom performance related to air cleanliness by particle concentration. Geneva: International Organization for Standardization; 2015.
40. Ministry of Health, Labour and Welfare (JP). Guidance on the manufacture of sterile pharmaceutical products produced by terminal sterilization. Tokyo: Ministry of Health, Labour and Welfare; 2012[acesso 10 maio 2020]. Disponível em: http://www.pmda.go.jp/files/000160794.pdf
41. Peyton TH, Abdou OA. Environmental control concepts for industrial clean-room facilities. J Archit Eng. 1995;1(1):53-63. http://doi.org/10.1061/(ASCE)1076-0431(1995)1:1(53)
42. International Organization for Standardization – ISO. ISO 14644 cleanrooms and associated controlled environments: part 2: specifications for testing and monitoring to prove continued compliance with ISO 14644-1. Geneva: International Organization for Standardization; 2000.
43. Pacheco FLC, Pinto TJA. The bacterial diversity of pharmaceutical clean rooms analyzed by the fatty acid metyl ester technique. PDA J Sci and Tech. 2010;64(2):156-66.
44. Sandle T. A review of cleanroom microflora: types, trends and patterns. PDA J Sci and Tech. 2011;65(4):392-403. http://doi.org/10.5731/pdajpst.2011.00765
45. Andon BM. Active air vs passive air (settle plate) monitoring in routine environmental monitoring programs. PDA J Sci and Tech. 2006;60(6):350-5.
46. Pasquarella C, Pitzurra O, Savino A. The index of microbial a contamination. J Hosp Infect. 2000;46(4):241-56. http://doi.org/10.1053/jhin.2000.0820
47. Sandle T. Settle plate exposure under unidirectional airflow and the effect of weight loss upon microbial growth. Eur J Parent Pharm Sciences. 2015;20(2):45-50.
48. Parenteral Drug Association – PDA. Fundamentals of an environmental monitoring program: technical report no 13: revised: parenteral drug association. PDA J Pharm Sci Technol. 2015;55(5 supl.TR13):1-35.
49. Japanese Pharmacopeia. Microbiological environmental monitoring methods of processing areas for sterile pharmaceutical products. Tokyo: Japanese Pharmacopeia; 2016.
50. Sandle T, Vijayakumar R, Al Aboody MS, Saravanakumar S. In vitro fungicidal activity of biocides against pharmaceutical environmental fungal isolates. J Appl Microbiol. 2014;117(5):1267-73. http://doi.org/10.1111/jam.12628
51. The United States Pharmacopeia – USP. Microbiological evaluation of clean rooms and other controlled environments. 39a ed. Rockville: The United States Pharmacopeial; 2011.
52. International Organization for Standardization – ISO. ISO 14644 cleanrooms and associated controlled environments: part 1: classification of air cleanliness. Geneva: International Organization for Standardization; 2015a.
53. Krippner E. Classificação de áreas limpas. Pharma Arquit. 2009: 42-5.
Autor notes
Ramos MJ – Concepção, planejamento (desenho do estudo), aquisição, análise, interpretação dos dados e redação do trabalho. Rito PN, Vieira W – Concepção, planejamento (desenho do estudo) e redação do trabalho. Todos os autores aprovaram a versão final do trabalho.
* E-mail: marcellejacomelli@hotmail.com
Declaração de interesses
Os autores informam não haver qualquer potencial conflito de interesse com pares e instituições, políticos ou financeiros deste estudo.