ARTIGO
Recepção: 28 Agosto 2020
Aprovação: 09 Março 2021
DOI: https://doi.org/10.7440/res64.2018.03
RESUMO
Introdução: A dipirona é um potente analgésico e antipirético inscrito na Relação Nacional de Medicamentos Essenciais. Ademais é estrategicamente indicada em casos especiais, como no tratamento paliativo de: dengue, chikungunya, zika e COVID-19. Problemas pós-comercialização, como não conformidades na qualidade, vêm sendo apontados para medicamentos contendo dipirona, resultando em recolhimentos frequentes pela Agência Nacional de Vigilância Sanitária.
Objetivo: Averiguar a qualidade e a equivalência farmacêutica dos comprimidos de dipirona, referência, genéricos e similares, disponíveis comercialmente para a população da cidade de Salvador/BA.
Método: Estudo experimental utilizando metodologias descritas na monografia da dipirona sódica comprimidos da Farmacopeia Brasileira 6ª edição. Foram analisadas três diferentes marcas de genérico, dois similares e o referência, coletadas no comércio da cidade de Salvador-Bahia. Adicionalmente, foi desenvolvido um método analítico alternativo por espectrofotometria na região ultravioleta para a análise da dipirona sódica comprimidos, validado com base na RDC n° 166, de 24 de julho de 2017.
Resultados: Os comprimidos de dipirona sódica referência, genéricos e similares atenderam aos requisitos quanto aos testes de identificação e características. Uma amostra de medicamento similar não atendeu ao teste de dissolução e somente duas amostras, dos medicamentos genéricos, foram satisfatórias para a intercambialidade. O método farmacopeico para doseamento da dipirona sódica comprimidos demonstrou problemas relacionados à subjetividade do analista e a incerteza do método. O método alternativo proposto mostrou-se seletivo, linear, preciso, exato e robusto, indicado para a quantificação da dipirona em comprimidos.
Conclusões: O monitoramento pós-comercialização de medicamentos com elevado padrão de consumo pela população, como a dipirona, pode sinalizar questões a serem investigadas pela vigilância sanitária, com o objetivo de apoiar sua missão de proteção à saúde da população.
Palavras chave: Dipirona, Controle de Qualidade, Perfil de Dissolução, Método Analítico, Validação.
ABSTRACT
Introduction: Dipyrone is a potent analgesic and antipyretic registered in the Brazilian List of Essential Medicines. Furthermore, it is strategically indicated in special cases, such as the palliative treatment of dengue, chikungunya, zika and COVID-19. Post-marketing problems have been identified for this drug, such as quality non-conformities, resulting in frequent withdrawals of medicines containing dipyrone by Anvisa.
Objective: To investigate the quality and pharmaceutical equivalence of dipyrone tablets (reference, generics and similar), commercially available in Salvador-Bahia.
Method: An experimental study was carried out using methodologies described in the Brazilian Pharmacopoeia 6th edition monograph of sodium dipyrone tablets. Three different brands were analyzed (one generic, two similar and one reference), all collected in Salvador-Bahia. In addition, an alternative analytical method by UV spectrophotometry was developed for the analysis of sodium dipyrone tablets, validated based on RDC No. 166/17.
Results: The reference, generic and similar sodium dipyrone tablets met the requirements for identification and characteristics tests. A similar drug sample did not meet the dissolution test and only two generic samples were satisfactory for interchangeability. The assay pharmacopoeial method for dipyrone sodium tablets showed problems related to the analyst’s subjectivity and method uncertainty. The proposed alternative method proved to be selective, linear, precise, accurate, and robust, and indicated for the quantification of dipyrone in tablets.
Conclusions: From the results obtained, it was observed that post-marketing monitoring of medicines with a population high consumption pattern, such as dipyrone, can signal issues to be investigated by health surveillance, with the objective of supporting its mission that is to protect population’s health.
Keywords: Dipyrone, Quality Control, Dissolution Profile, Analytical Method, Validation.
INTRODUÇÃO
A dor e a febre fazem parte dos cincos sinais cardinais da inflamação, que consistem em uma rede de respostas complexas à lesão tecidual e infecções1 . Um dos medicamentos mais utilizados para o tratamento da dor e da febre é a dipirona (DIP)2 , 3 , da classe dos anti-inflamatórios não esteroidais.
A DIP também é estrategicamente indicada na substituição do ácido acetilsalicílico, principalmente, em situações em que este não é recomendado, como é o caso do tratamento de arboviroses, a exemplo da: dengue, zika e chikungunya2 . Mais recentemente, reforçando a sua relevância, a DIP foi elencada como recurso terapêutico para a COVID-19, transmitida pelo novo coronavírus (SARS-CoV-2), infecção considerada pela Organização Mundial da Saúde, em março de 2020, uma pandemia, devido a sua magnitude4 , 5 , 6 .
Ainda, a DIP consta na Relação Nacional de Medicamentos Essenciais (Rename), como Componente Básico da Assistência Farmacêutica, compondo os medicamentos que devem atender às necessidades de saúde prioritárias da população brasileira no Sistema Único de Saúde7 .
Entretanto, embora a DIP tenha a sua efetividade comprovada e apresente boa relação risco-benefício, dados adicionais são necessários para assegurar, também, a qualidade das formulações e garantir a segurança e o sucesso terapêutico, considerando que a DIP é um medicamento isento de prescrição e o seu consumo no Brasil só vem aumentando8 .
Atrelado a esse fato, a DIP está presente na lista de medicamentos candidatos à bioisenção, ou seja, isentos de estudos de biodisponibilidade relativa/bioequivalência ( in vivo ), desde que o medicamento que o contenha atenda aos requisitos de qualidade previstos nos ensaios in vitro9 , 10 , 11 . Neste caso, o perfil de dissolução passa a ser fundamental para prever o desempenho in vivo do medicamento e, portanto, para a comprovação da sua segurança e eficácia farmacológica9 , 11 , parâmetros correspondentes à intercambialidade dos medicamentos referência, genérico e similar.
Junto ao exposto, a ocorrência frequente de recolhimentos pela Agência Nacional de Vigilância Sanitária (Anvisa) denota que a qualidade dos medicamentos pode apresentar não conformidades. No tocante à DIP, a Anvisa relata o recolhimento pós-comercialização devido a desvios de qualidade12 , 13 , os quais podem gerar riscos à saúde e aumentar o número de hospitalizações, elevar os gastos públicos com a saúde, além de lesar o direito do consumidor14 , 15 .
Isto posto, justifica-se a necessidade e objetivou-se avaliar a qualidade, a equivalência farmacêutica e o perfil de dissolução da DIP sódica comprimido em medicamentos de referência, genéricos e similares na pós-comercialização, cidade de Salvador/BA, como forma de indicar se os medicamentos comercializados estão dentro dos padrões de qualidade e de intercambialidade aceitáveis.
MÉTODO
A substância química de trabalho (SQT), lote 15720, validade 01/2019 e teor 99,5%, foi gentilmente cedida pelo Núcleo Controle de Qualidade da Universidade Federal de Alfenas. Todos os solventes e reagentes utilizados foram de grau analítico. As amostras analisadas representam comprimidos simples de DIP sódica contendo 500 mg de ativo (teor declarado), classificados e codificados como medicamentos de referência (R), genéricos (G1, G2 e G3) e similares (S1 e S2) (n = 90 unidades de cada), adquiridos em farmácias comerciais da cidade de Salvador, na Bahia, no ano de 2018. Todos os comprimidos se apresentavam brancos, cilíndricos e com diâmetro de aproximadamente 0,13 mm. Segundo as bulas das amostras avaliadas, além da DIP monoidratada, R foi composto por estearato de magnésio – Mg(C18H35O2)2 – e macrogol 4.000; G1 e G3 por celulose microcristalina, fosfato de cálcio dibásico dihidratado, povidona, edetato dissódico (EDTA) e Mg(C18H35O2)2; G2 foi composto por croscarmelose monoidratada, Mg(C18H35O2)2, dióxido de silício (SiO2), sacarose e amido; S1 por hipromelose, povidona, SiO2, sacarose, amarelo de quinolina, amido, talco, Mg(C18H35O2)2, álcool etílico e água purificada; e S2 por celulose microcristalina PH 101, lactose monoidratada, polivinilpirrolidona (PVP), Mg(C18H35O2)2, talco, amido, álcool etílico e água purificada.
Todo o delineamento experimental dessa pesquisa foi realizado com as amostras dentro do prazo de validade e baseado na 6ª edição da Farmacopeia Brasileira (FB 6)16 , conforme a monografia individual de DIP monoidratada comprimidos (EF124-00), além das Resoluções da Diretoria Colegiada (RDC) n° 166, de 24 de julho de 201717 , e RDC n° 31, de 11 de agosto de 20109 .
Quanto aos equipamentos, foi utilizada balança analítica Bioprecisa (modelo FA2104N), banho de ultrassom Cristófoli, desintegrador Nova Ética (modelo 301/AC01), dissolutor Ethik (modelo 299/3), durômetro Ethik (modelo 298/DGP) e espectrofotômetro Shimadzu (modelo UV1800).
Identificação
As avaliações físico-químicas englobaram os testes de identificação, A e B, a partir dos comprimidos pulverizados ( pool ), reagindo com peróxido de hidrogênio concentrado e persulfato de potássio a 10% (p/v)16 , respectivamente.
Características
As características dos comprimidos de DIP analisados incluíram determinação de peso, dureza e friabilidade, teste de desintegração e uniformidade de doses unitárias, os quais seguiram os métodos gerais e especificações da FB 616 .
Teste de dissolução
Seguindo a especificação da monografia da DIP comprimido16 , utilizou-se como meio de dissolução ácido clorídrico (HCl) 0,1 M, com volume de 500 mL, aparato pá, velocidade de 50 rpm e tempo do teste de 45 min. Após os testes foram retiradas alíquotas do meio de dissolução das seis cubas, as quais foram filtradas e diluídas em HCl 0,1 M até a concentração de 20 µg/mL. As absorvâncias das soluções foram determinadas por método espectrofotométrico na região do ultravioleta (UV), a 258 nm, utilizando o mesmo solvente para ajuste do zero. Calculou-se a quantidade de DIP sódica dissolvida pela equação da reta da curva analítica de DIP SQT. A tolerância farmacopeica indica que não menos do que 70% (Q) da quantidade declarada de DIP se dissolve em 45 min.
Perfil de dissolução
Para o estabelecimento do perfil de dissolução foram utilizados doze comprimidos de cada amostra (n = 12). Seguiram-se todas as especificações de dissolução e análise descritas para o teste de dissolução. As alíquotas foram retiradas do meio de dissolução nos tempos 5, 10, 15, 20, 25, 35 e 45 min, filtradas e diluídas em HCl 0,1 M até a concentração de 20 µg/mL. A avaliação dos perfis foi feita por meio da curva de porcentagem dissolvida versus o tempo e pelo Método Modelo Independente Simples, o qual emprega um fator de diferença (F1) e um fator de semelhança (F2). Para a intercambialidade, os medicamentos comparados devem apresentar tipos de dissoluções correspondentes e valor de F2 compreendido entre 50 a 1009 .
Cinética e Eficiência de Dissolução (ED%)
Quatro modelos matemáticos foram aplicados para avaliar a cinética de liberação nas especialidades estudadas: ordem zero (com equação Qt = Q0 − K0t , no qual o gráfico da fração do fármaco não dissolvido versus o tempo é linear), primeira ordem (1n Qt = 1n Q0 − K1t , cujo gráfico do logaritmo neperiano da quantidade não liberada do fármaco versus o tempo é linear), Higuchi ( ft = KHt1, para o qual o gráfico da fração do fármaco dissolvido versus a raiz quadrada do tempo é linear) e Hixson-Crowell ( W01 − Wt1 = Kst , cujo gráfico da raiz cúbica da fração não liberada do fármaco versus o tempo é linear). Para as equações dos modelos cinéticos descritos anteriormente, Qt é a quantidade de fármaco dissolvida no tempo t ; Q0 é a quantidade inicial de fármaco na solução; K0 e K1 são as constantes de liberação de ordem zero e primeira ordem, respectivamente; ft é a quantidade de fármaco liberado no tempo t por unidade de superfície; KH é a constante de dissolução de Higuchi; W0 é a quantidade inicial de fármaco na forma farmacêutica; Wt é a quantidade de fármaco que permanece na forma farmacêutica no tempo t ; Ks é a constante que incorpora a relação superfície-volume18 .
As curvas foram construídas considerando-se somente os pontos que correspondiam à fase ascendente da liberação do fármaco, ou seja, até 20 min para o R, até 30 min para os medicamentos G1 e G3, e até 45 min para os demais, a saber, G2, S1 e S2. O modelo matemático que, até o fim do experimento (45 min), melhor expressou o perfil de dissolução da DIP comprimidos em cada amostra (n = 12) foi selecionado com base no coeficiente de correlação (r)19 , por meio da linearização dos perfis de dissolução.
Dados adicionais, mais detalhados, também foram estabelecidos: constante de velocidade de dissolução (k), a qual corresponde ao coeficiente angular da inclinação da reta de regressão, meia-vida de dissolução (t50%), conceituada como o tempo no qual se dissolvem 50% de DIP, e quantidade dissolvida em 30 min (Q30), que retrata a concentração do ativo no ponto máximo de uma dissolução caracterizada como rápida10 .
Por fim foi calculada a ED%, a partir da razão dos valores de área sob a curva (ASC0 45) do perfil de dissolução de DIP, nas diferentes amostras, em um intervalo de tempo de 45 min, pelo método dos trapezoides20 . A área total do retângulo (ASCTR) foi definida pela ordenada 100% de dissolução e pela abscissa tempo igual a 45 min.
Os valores de ED% obtidos foram submetidos à análise de variância one-way (ANOVA), seguidos de sua comparação pelo teste de Tukey.
Doseamento por titulometria
Para a determinação do teor de DIP nas amostras de comprimidos, 20 unidades foram pesadas e pulverizadas. A partir desse pó ( pool ), quantidade equivalente a 0,35 g de DIP sódica foi pesada, transferida e dispersada em um erlenmeyer contendo 25 mL de água e 5 mL de ácido acético glacial. A titulação foi realizada com iodo 0,05 M (SV), em temperatura abaixo de 15°C, utilizando 1 mL de amido (Solução Indicadora), como indicador, sendo a visualização do ponto final feita por mudança de cor. Cada mL de iodo 0,05 M SV equivale a 17,570 mg de DIP. A especificação farmacopeica para este teste é de, no mínimo, 95,0% e, no máximo, 105,0% da quantidade declarada de DIP sódica16 .
Desenvolvimento e validação do método por espectrofotometria
Foram preparadas quatro soluções-mãe pesando-se, exatamente, 10 mg de DIP SQT. As massas do ativo foram transferidas para quatro balões volumétricos (BV) de 100 mL e avolumou-se cada BV com o respectivo solvente, a saber: água, HCl 0,1 M, hidróxido de sódio (NaOH) 0,1 M e metanol, obtendo-se concentração de 100 μg/mL. Posteriormente, realizou-se a diluição para a concentração de 20 μg/mL. A partir destas soluções efetuaram-se as varreduras no espectrofotômetro entre os comprimentos de onda de 400 e 200 nm.
Para a validação analítica do método proposto foram avaliados oito parâmetros: 1) seletividade, pela comparação dos espectros de absorção no UV de soluções de DIP SQT, amostra (R) e placebo (estearato de magnésio e macrogol 4000), utilizando HCl 0,1 M como solvente; 2) intervalo, nas concentrações de 1, 3, 5, 8, 10, 12, 15, 18, 20, 25, 30, 40 e 50 μg/mL, analisadas pela curva de Ringbom; 3) linearidade, pela construção da curva analítica final, em seis concentrações, 5, 10, 15, 20, 25 e 30 μg/mL, avaliando-se a representação gráfica das absorvâncias obtidas em função da concentração da DIP; o gráfico de dispersão dos resíduos, acompanhado de sua análise estatística; a equação da reta de regressão de y em x, estimada pelo método dos mínimos quadrados; a avaliação da associação linear entre as variáveis por meio dos coeficientes de correlação (> 0,990) e de determinação (R2) e a avaliação da significância do coeficiente angular. O teste de Anderson-Darling foi utilizado para a avaliação da normalidade dos resíduos, como requerido pela RDC n° 166/201717 . A homocedasticidade dos dados também foi investigada para a utilização do modelo adequado. Nos testes estatísticos foi utilizado nível de significância de 5%; 4) precisão , avaliada por seis réplicas, na concentração de trabalho, 20,0 μg/mL, nas mesmas condições de operação, mesmo analista e mesma instrumentação, em uma única corrida analítica, para a avaliação da precisão intradia (repetibilidade) e no mesmo laboratório, em pelo menos dois dias diferentes, realizada por operadores distintos, para a determinação da precisão interdias (precisão intermediária). Os resultados foram determinados pela dispersão dos valores, calculando-se o desvio-padrão relativo (DPR%) da série de medições; 5) exatidão , avaliada pelo método de adição de padrão, no qual quantidades conhecidas de SQT (6, 10, e 14 μg/mL) foram acrescidas à solução do produto acabado (10 μg/mL), em três diferentes concentrações da faixa de uso do método (baixa, média e alta – 16, 20 e 24 μg/mL), com leituras em triplicata; 6) limite de detecção (LD) e 7) limite de quantificação (LQ), ambos estimados pelos parâmetros da curva de analítica, valor da inclinação (IC) e o desvio padrão residual da linha de regressão (s); e 8) robustez, pelo planejamento fatorial de Youden21 , sendo efetuadas variações (condição normal e alterada, nesta ordem) na molaridade do solvente (0,1 M e 0,09 M), no tempo de ultrassom (30 min e 15 min), no analista (1 e 2), no comprimento de onda (258 e 259 nm), no dia de preparo da solução-mãe (dia da análise e 24 h anteriores – solução-mãe mantida sob refrigeração), no tamanho do balão volumétrico (10 e 5 mL) e na marca da micropipeta (Peguepet® e Rainin®).
Todos os parâmetros foram avaliados de acordo com os documentos do International Conference on Harmonisation22 , do Instituto Nacional de Metrologia, Qualidade e Tecnologia23 e da RDC nacional n° 166/201717 , os quais dispõem sobre a validação de métodos analíticos. Para os testes que necessitaram da utilização do produto acabado foi utilizado o medicamento R.
Doseamento por espectrofotometria na região do UV
Para o doseamento das amostras de DIP em comprimidos, pelo método espectrofotométrico na região do UV, foram pesados, a partir do pool , o equivalente a 10 mg de DIP de cada amostra. Cada massa pesada foi transferida para BV de 100 mL, acrescida da solução de HCl 0,1 M e submetida a banho ultrassônico por 30 min. Após filtração em papel de filtro quantitativo, foram retiradas alíquotas e preparadas soluções de trabalho na concentração de 20 μg/mL. As soluções foram analisadas, de acordo com o método desenvolvido e validado, no comprimento de onda de 258 nm, utilizando HCl 0,1 M como solvente e branco.
RESULTADOS E DISCUSSÃO
É indispensável a análise da qualidade dos medicamentos já disponíveis no mercado, pois, sem a garantia de que os fármacos e medicamentos disponíveis apresentam padrões de qualidade, segurança e eficácia aceitáveis, qualquer serviço de saúde fica prejudicado18 .
Em relação aos ensaios de verificação da qualidade, o presente trabalho apresentou os resultados expostos a seguir.
Identificação
Via de regra, análises utilizando reações químicas não visam identificar toda a molécula do fármaco, mas grupos funcionais ou determinados grupos químicos importantes. No caso da DIP, nos testes A e B, sugere-se que aconteçam quebras de ligações orgânicas (nos grupos hidrazina quinolínico e pirazol), possíveis oxidações, com formação de espécies de transição, liberação de gás (O2) e calor (reação exotérmica). A partir dos resultados obtidos, pode-se afirmar, de forma qualitativa, que todas as amostras têm presentes em sua composição grupamentos químicos que são característicos do princípio ativo DIP.
Características
O teste de determinação de peso permite verificar se as unidades de um mesmo lote apresentam uniformidade de peso e, consequentemente, refletem sobre a adequada fabricação dos comprimidos.
Os resultados do teste de determinação de peso para os comprimidos de DIP, formulações R, G1, G2, G3, S1 e S2 estão apresentados na Tabela 1 . A variação de peso aceitável para comprimidos com peso médio igual ou superior a 250 mg é de ± 5%16 . Apenas um dos comprimidos do medicamento S1 apresentou-se abaixo do limite inferior. Porém, como é aceitável até dois comprimidos fora do limite estabelecido, aponta-se não haver não conformidades para os lotes avaliados nesse teste. Isso demonstra a homogeneidade de peso, e consequentemente, regularidade da produção, sugerindo bom fluxo da mistura de pós no momento da fabricação dos lotes analisados15 .
Resultados dos testes de determinação de peso, dureza, desintegração, friabilidade, uniformidade de doses unitárias e doseamento para os comprimidos simples de dipirona (amostras R, G1, G2, G3, S1 e S2).
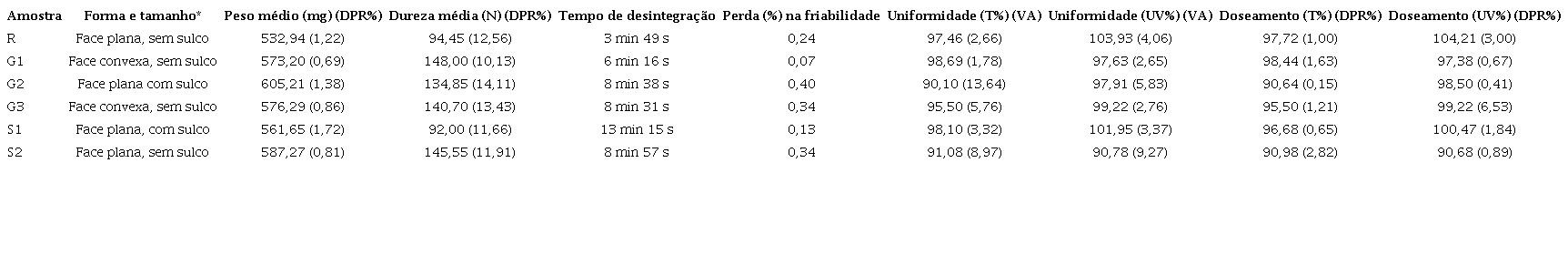
*Todas as amostras estudadas foram de comprimidos simples de liberação imediata, brancas, cilíndricas e com diâmetro de aproximadamente 0,1303 mm. N: Newtons; DPR%: Desvio padrão relativo; T%: Média do teor calculado/encontrado utilizando o doseamento por titulação; UV%: Média do teor calculado/encontrado utilizando o doseamento por espectrofotometria na região do UV; VA: Valor de aceitação.
Estatisticamente, todas as amostras apresentaram diferença, quando comparadas ao peso médio obtido para o medicamento R ( p -valor < 0,05). Este resultado já era esperado, posto que, qualitativamente, as amostras analisadas apresentaram excipientes substancialmente distintos. G1 e G3 apresentaram a mesma composição e pesos ( p -valor = 0,7113).
Testes de determinação de peso realizados com comprimidos de DIP em outros municípios brasileiros também não apresentarem divergências quanto a homogeneidade do peso de suas amostras8 , 24 , 25 .
Para os ensaios de resistência mecânica, foram realizados os testes de dureza e friabilidade. O teste de dureza é um ensaio que proporciona a determinação da resistência do comprimido, o qual, segundo a FB 616 , é de caráter informativo. Dentro dos resultados obtidos, somente a amostra S1 não diferiu do medicamento R ( p -valor = 0,9995). Entretanto, foram percebidos valores com alta variabilidade na força necessária à ruptura dos comprimidos (DPR% de 10,13 a 14,11%), indicando possíveis problemas na produção.
O teste de friabilidade é um teste importante, que visa observar o desprendimento de partículas do produto, permitindo determinar a resistência dos comprimidos à abrasão16 . Com os resultados encontrados, podemos afirmar que as amostras dos comprimidos de DIP, após serem submetidos ao teste, apresentaram baixa porcentagem de perda de pós para os medicamentos referência, genéricos e similares, com perda igual ou inferior a 1,5% do seu peso16 . Adicionalmente, os valores do teste friabilidade corroboraram os resultados do teste de dureza. Via de regra, quanto maior é a força de compressão aplicada e, portanto, a dureza, menor é a friabilidade, embora outras características, também afetem a resistência mecânica dessa forma farmacêutica.
No trabalho feito por Lanna et al.24 , os comprimidos de DIP referência, genérico e similar apresentaram-se em não conformidade, em relação à friabilidade, sendo os similares responsáveis pela maior perda de massa (acima de 6%), o que pode incluir também perda de princípio ativo. Essas diferenças nos resultados mostraram que lotes diferentes em épocas distintas podem implicar em variações na qualidade, o que reforça a importância de análises pós-comercialização dos medicamentos.
Usualmente, os comprimidos devem ser suficientemente duros, para resistir à ruptura durante os processos a que estão sujeitos, e frágeis o bastante para sofrer desintegração no tempo adequado, após a ingestão26 .
Neste estudo foram observados tempos de desintegração, para todas as amostras, dentro do tempo máximo permitido para comprimidos não revestidos (30 min)16 . Entretanto, como afirma Cazedey18 , uma rápida desintegração não significa que o fármaco será absorvido, por outro lado, a avaliação deste parâmetro é significativa, uma vez que, se a desintegração não ocorrer, provavelmente, o fármaco não poderá ser absorvido.
Para garantir um tratamento adequado, cada comprimido do lote de um medicamento deve conter quantidade do componente ativo próxima da quantidade declarada pelo fabricante. O teste de uniformidade de dose unitária permite assegurar a administração de doses corretas e homogêneas, por meio dessa verificação16 .
Os resultados encontrados para os valores de aceitação, variaram de 1,78 a 13,64. À vista disso, todos os medicamentos testados mostraram-se em conformidade com a especificação (VA < 15,0), o que garante a homogeneidade e uniformidade de distribuição da DIP nas doses individuais.
Teste de dissolução
O ensaio de dissolução pode ser entendido como um teste in vitro que simula o comportamento do medicamento in vivo . A dissolução pode ser dita como um processo no qual um fármaco é liberado da sua forma farmacêutica e se transforma em forma livre para ser absorvido pelo organismo28 .
De acordo com a Tabela 2 , os resultados para as amostras R, G1, G2, G3 e S1 são satisfatórios, uma vez que cada unidade apresenta valor de dissolução acima de 75% (Q + 5%) no tempo de 45 min, atendendo assim ao preconizado para o teste de dissolução de comprimidos de DIP já no estágio E116 . Entretanto, os resultados de dissolução da amostra S2, para todas as unidades, apresentaram valores de liberação inferiores ao requerido. Esse resultado já era esperado, uma vez que essa amostra apresentou valores de doseamento abaixo de 95%16 , em desacordo com o requisito farmacopeico. Para o medicamento S2 unidades adicionais foram testadas, estágio E2 e E3, porém, os resultados não foram satisfatórios. Não conformidades em medicamentos similares para esse teste foram encontradas, semelhantemente, na literatura24 , 25 .
Resultado do teste de dissolução para os comprimidos de dipirona.
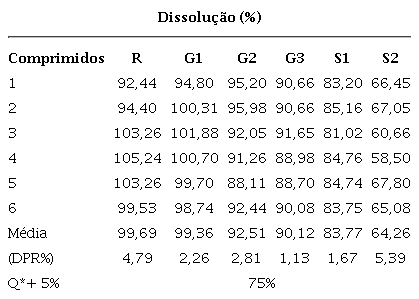
*Q (quantidade dissolvida de fármaco) determinada pela FB 616 de 70% em 45 min.
Perfil de dissolução
Para o perfil de dissolução, efetuam-se coletas em múltiplos pontos, obtendo-se a representação real do processo de dissolução. O perfil de dissolução permite uma análise mais conclusiva da liberação do ativo e é muito empregado para a demonstração da semelhança ou diferença entre medicamentos teste e referência, sob todas as condições testadas9 .
Na Figura 1 podem ser verificados os perfis de dissolução dos medicamentos testados, ou seja, a porcentagem de princípio ativo sendo liberado em função do tempo.
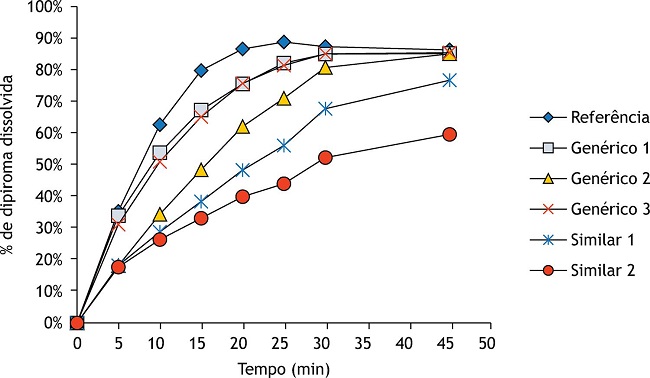
Figura 1
Perfis de dissolução dos comprimidos de dipirona (amostras R, G1, G2, G3, S1 e S2).
Fonte: Elaborada pelos autores, 2020.*Nota: os primeiros três pontos de coleta para cada não excederam 20% de variação. Os demais pontos ficaram abaixo de 10%.
Como esperado para compostos altamente solúveis (Classe I29 - Sistema de Classificação Biofarmacêutica), a dissolução da DIP no medicamento R mostrou-se rápida, isto é, apresentou valor igual ou superior a 85% de fármaco dissolvido em até 30 min9 . Dessa forma, os medicamentos candidatos à equivalentes deveriam exibir o mesmo perfil. Entretanto, como pode ser visualizado na Figura 1 , apenas as amostras G1 e G3 apresentaram tipos de dissoluções correspondentes à dissolução do medicamento R.
Ainda pode ser analisado que o medicamento G2, apesar de atender à especificação de liberação (teor) em 45 min, apresentou perfil de dissolução mais lento, sempre comparado ao R. Assim, é relevante salientar que a amostra G2 também teria sido considerada adequada, se fosse julgado somente o ensaio de dissolução propriamente dito ou de um ponto. S1 e S2 diferiram completamente da amostra R.
A variação ponto a ponto entre as amostras analisadas, as quais apresentaram DRP (%) de 33,40, 34,96, 33,01, 24,53, 17,99 e 13,15, correspondendo, respectivamente, aos pontos, 5, 10, 15, 20, 25, 30 e 45 min corroboram a validade do teste de perfil de dissolução, uma vez que suprime o erro de avaliação somente pelo ponto final de dissolução.
Outro aspecto importante a ser notado é que nenhuma amostra obteve 100% de liberação do ativo no tempo definido pela Farmacopeia Brasileira16 para o ensaio, provavelmente devido à agitação mecânica, que, para o teste de doseamento, é mais eficiente (ultrassom) que a energia hidrodinâmica exercida pelo aparato pá do teste de dissolução.
Por fim, posto que a DIP é um medicamento candidato a bioisenção e, considerando que o conceito de Equivalentes Farmacêuticos engloba “medicamentos que possuem mesma forma farmacêutica, mesma via de administração e mesma quantidade da mesma substância ativa, isto é, mesmo sal ou éster da molécula terapêutica, podendo ou não conter excipientes idênticos, desde que bem estabelecidos para a função destinada”9 , no que tange o papel dos excipientes contidos nos medicamentos G2, S1 e S2, era previsto que os mesmos não interferissem na liberação do ativo, o que não foi confirmado, pelo contrário, aparentemente, a presença dos diferentes coadjuvantes exerceu papel controlador da dissolução.
Resultado semelhante foi obtido na determinação dos valores de F1 e F2. Como pode ser visto na Tabela 3 , somente as amostras G1 e G3 apresentaram resultados de F1 menores que 15 e de F2 entre 50 a 100, ou seja, satisfatórios para a intercambialidade.
Parâmetros calculados pelo estudo da cinética de dissolução de dipirona comprimidos.
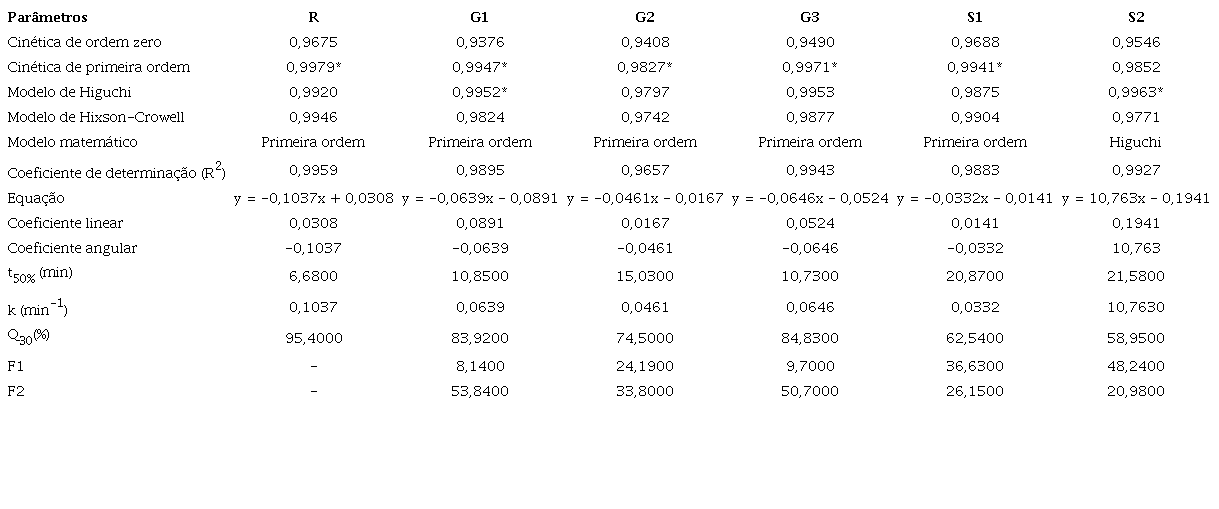
*Modelos com melhor correlação; Especificações: F1 < 15 e F2 entre 50 a 10013.
Esse comportamento diferenciado das amostras de genéricos e similares em relação ao medicamento referência também já foi identificado em outros estudos com a DIP comprimidos e revela a importância da avaliação do perfil de dissolução para o monitoramento pós-comercialização dos medicamentos24 , 25 .
Cinética e ED
Conforme os resultados mostrados na Tabela 3 , observou-se que o modelo mais adequado, isto é, aquele que forneceu maiores valores de r para os medicamentos R, G2, G3 e S1, foi o de primeira ordem. A equação de primeira ordem delineia o sistema de liberação no qual a taxa de dissolução é dependente da concentração das espécies dissolvidas30 .
Para a amostra G1, os valores de correlação obtidos pela linearização dos perfis de dissolução nos modelos matemáticos de primeira ordem e Higuchi mostraram-se muito próximos e, considerando o tipo de dissolução dessa amostra, optou-se por classificá-lo como de primeira ordem.
Já para o medicamento S2, o melhor modelo para a predição da liberação da DIP, indubitavelmente, foi o de Higuchi. A equação de Higuchi apresenta o sistema de liberação em que o fármaco sólido é disperso em uma matriz insolúvel e a taxa de liberação do fármaco é relacionada com a sua taxa de difusão31 , 32 . A classificação do medicamento S2 neste modelo matemático explica muito a sua baixa performance na liberação da DIP e alerta para a verificação da formulação farmacêutica.
A Tabela 3 também apresenta os parâmetros calculados pelo estudo da cinética de dissolução da DIP comprimidos nas amostras. Os resultados de Q30 corroboram os tipos de dissolução10 . Os valores de k, embora não representem muito em si, são importantes, pois são empregados nos cálculos de t50%. Este, por sua vez, é de extrema importância, uma vez que reflete a velocidade de liberação. Assim, para produtos nos quais a liberação do fármaco é mais rápida, como nas amostras R, G1, G2 e G3, os valores de t50% serão mais baixos, de até, aproximadamente, 15 min. Esta observação sugere que, em um curto espaço de tempo, a forma farmacêutica libera metade da quantidade do fármaco, fato realmente interessante para uma forma farmacêutica que contém um fármaco classe I29 e que requer ação terapêutica rápida.
Contudo, mesmo entre os medicamentos em estudo que obedeceram a mesma cinética (primeira ordem), observou-se diferenças importantes. O produto R apresentou a maior velocidade de dissolução, seguido pelos produtos G1 e G3, os quais comportaram-se de forma semelhante, apresentando valores análogos para k e bastante próximos para t50%. O produto G2 demonstrou dissolução mais lenta, sendo mais vagarosa a velocidade de dissolução da amostra S1. Para o medicamento S2 todos os parâmetros cinéticos calculados demostraram inconsistências.
As variações observadas podem ser explicadas por diferenças nas matérias-primas, formulações, processos de fabricação, condições de transporte e armazenamento e devem ser investigadas.
Por fim, foi avaliada a ED%, um parâmetro de confiança que pode ser utilizado para efetuar a comparação entre formulações e permite, segundo alguns autores, comparação mais fidedigna e detalhada entre dois produtos ou situações20 , 33 , 34 , 35 . Além disso, a ED% apresenta a vantagem de poder ser relacionada com a quantidade real de fármaco que se encontra dissolvida no organismo, uma vez que a extensão da absorção de um fármaco in vivo é proporcional à sua concentração dissolvida e ao tempo que permanece em contato com as regiões de absorção34 , 36 .
Os valores de ED% encontrados para as amostras R, G1, G2, G3, S1 e S2 foram 84,60%, 79,67%, 68,64%, 79,02%, 63,75% e 66,30%, respectivamente.
Obteve-se o p -valor de 1,407 x 10- 14 , pela ANOVA, demonstrando que existem diferenças significativas entre os medicamentos em relação à ED%. Os resultados obtidos com o teste de Tukey indicam, corroborando os resultados obtidos anteriormente, que os valores de ED% são semelhantes para as amostras G1 e G3 e diferentes os medicamentos G2, S1 e S2, em relação ao medicamento R.
Doseamento por titulometria
Para o doseamento da DIP, a FB 616 indica a volumetria iodimétrica, a qual é baseado na oxidação do grupo metanossulfônico da DIP a sulfato em meio ácido.
Por ser um método farmacopeico, a titulometria foi utilizada para a determinação do teor de DIP nas amostras e obtiveram-se, como médias, os teores apresentados na Tabela 2 . A partir da análise dos resultados deste doseamento, percebeu-se que dois medicamentos não atenderam aos requisitos estabelecidos pela FB 616 de, no mínimo, 95,0% de teor do fármaco declarado. Isto implica que, para os lotes analisados, as amostras G2 e S2, mostraram teor abaixo de limite mínimo preconizado, podendo ocasionar falhas terapêuticas. Do mesmo modo, a literatura relata irregularidades em uma amostra de medicamento similar de DIP comprimidos, a qual apresentou valor médio de 89,7%27 .
Contudo, deve-se analisar que, durante a realização deste doseamento, alguns entraves foram encontrados. Primeiramente, notou-se uma dificuldade na verificação do ponto final da reação. Não havendo na FB 6 especificação do tempo de permanência da coloração azul. O controle da temperatura foi outra questão a ser considerada. A DIP sódica pode sofrer hidrólise, quando em solução aquosa e à temperatura ambiente. Por isto é tão importante realizar a titulação em temperatura abaixo de 15°C. Entretanto, alcançar esta condição de temperatura é trabalhoso e demorado, fazendo com que o método seja pouco prático. Diante das dificuldades apresentadas, os erros podem acabar sendo inseridos e, dessa forma, o método pode apresentar resultados pouco confiáveis.
Desenvolvimento e validação de método por espectrofotometria na região do UV
O desenvolvimento de um novo método para a DIP foi motivado por fatores relacionados a incerteza do método oficial para os ensaios de teor, bem como pela praticidade e indicação da própria farmacopeia para os ensaios de dissolução.
Desenvolvimento do método analítico por espectrofotometria na região do UV
Os resultados da varredura da DIP nos diversos solventes testados podem ser vistos na Figura 2 . Analisando-se os perfis de absorção de DIP obtidos nos quatros solventes utilizados, percebeu-se que a água, apesar de ser um solvente universal e apresentar diversas vantagens, demonstrou baixa sensibilidade. Com o NaOH 0,1 M, por sua vez, além da baixa sensibilidade percebeu-se muitos ruídos até aproximadamente 215 nm. Por fim, as soluções de DIP em HCl 0,1 M e metanol apresentaram os melhores picos de máxima absorção.
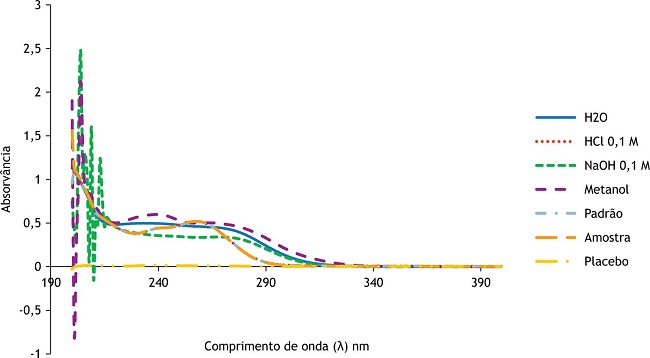
Figura 2
Varreduras na região do UV de dipirona em diferentes solventes e dipirona padrão, amostra e placebo em HCl 0,1 M.
Fonte: Elaborada pelos autores, 2020.
Concomitantemente, procedeu-se aos testes de taxa de extração (extração sólido-líquido), sendo obtidas as seguintes taxas da DIP, nos respectivos solventes e comprimentos de onda máximos: 99,90% com HCl 0,1 M, em 258 nm; 87,53%, com metanol, em 239 nm; 134,00% em NaOH 0,1 M, em 269 nm; e 89,45% em água em 233 nm. Isto posto, depreendeu-se que o solvente que apresentou a melhor taxa de extração da DIP a partir da matriz dos comprimidos foi o HCl 0,1 M.
Considerou-se, também, que o ácido pode ser reputado menos agressivo ao analista comparado ao metanol, o qual pode causar danos à saúde devido às suas propriedades narcóticas, de irritação das mucosas e de potencial cumulativo (produção de compostos tóxicos com baixa velocidade de eliminação), mesmo que em exposições curtas37 .
Validação
A validação foi realizada para demostrar que o método produz resultados confiáveis e é adequado para a quantificação da DIP23 .
A seletividade do método foi comprovada, como apresentado na Figura 2 , pela ausência de sinal na varredura do placebo, mostrando que não há interferência da matriz dos comprimidos na absorção da DIP e pela sobreposição dos perfis de absorção da molécula na SQT e na amostra, com ambos apresentando pico de absorção máxima no comprimento de onda de 258 nm, utilizando HCl 0,1 M como solvente.
O intervalo linear permitiu determinar a faixa de concentração em que o erro fotométrico relativo é mínimo39 , sendo as concentrações na inflexão da sigmoide obtida entre 5 e 30 µg/mL.
A curva analítica, construída no intervalo linear, demonstrou a forte correlação entre as concentrações da DIP e as absorvâncias obtidas, com equação da reta y = 0,0254x + 0,0044 e r e R2 iguais a 0,9999.
O p -valor do teste de Anderson-Darling de 0,6257 atesta a normalidade dos resíduos ao nível de significância de 5%. A significância do coeficiente angular foi verificada pelo teste F da ANOVA e, com p -valor igual a 0, entende-se que é significativamente diferente de zero. A homocedasticidade, com resultado de p -valor 0,092144, do Teste de Cochran, não rejeitou a hipótese de igualdade das variâncias ao mesmo nível de significância.
Na avaliação da precisão foram determinados, para a repetibilidade e a precisão intermediária, respectivamente, os valores de DPR de 0,47% e 1,67%, considerados baixos, de acordo com a Association of Official Analytical Chemists (AOAC)40 .
O parâmetro de exatidão do método foi comprovado pelos resultados de recuperação entre 97,42% e 102,22%, dentro do intervalo de 80,00% a 110,00%40 , para a concentração de 1 a 10 µg/mL.
Os valores para LD e LQ foram estimados em 0,041 e 0,125 μg/mL, presumindo a capacidade do método em detectar e quantificar baixas concentrações.
Por fim, a robustez, que fornece uma indicação da confiança do método durante uma aplicação rotineira, foi avaliada. Os resultados deste teste, por Youden, demonstraram que as variações estabelecidas para cada parâmetro apresentaram um efeito mínimo em todas as condições estudadas. Adicionalmente, foi utilizado o método matemático de Lenth, o qual resultou em p -valores menores que 0,05, rejeitamos a hipótese de efeitos ativos ao nível de significância de 5%.
Doseamento por espectrofotometria na região do UV
A Tabela 1 apresenta o doseamento obtido para cada amostra. Os medicamentos apresentaram valores médios entre 90,69% e 104,21%, apontando que a amostra S2 não apresentou teor dentro dos valores tolerados pela FB 616 .
Comparação dos métodos utilizados no doseamento
Analisando, comparativamente, os resultados obtidos com os dois métodos empregados no doseamento da DIP, podemos depreender que ambos, por titulometria e por espectrofotometria na região do UV, apresentaram resultados estatisticamente semelhantes para os medicamentos G1, G3, S1 e S2, com p -valores acima de 0,05 (0,4356, 0,4691, 0,0519 e 0,8848, respectivamente).
Entretanto, os resultados para as amostras R e G2 foram diferentes ( p -valores de 0,0486 e 1,31x10-5, nesta ordem). É importante analisar que a amostra R, apesar de apresentar teor dentro da especificação farmacopeica, mostrou resultados extremos com valores de 97,72% na avaliação com o método clássico e 104,21% na determinação com o método instrumental.
Ainda, o medicamento G2 apresentou-se não conforme no teste de doseamento, quando se empregou o método iodimétrico, no entanto, foi aprovado quando sua análise foi realizada com o método espectrofotométrico.
É válido ponderar também que a técnica de titulação consome mais reagente, mais tempo e apresenta a incerteza do analista (subjetividade), especialmente no que se refere ao ponto final da reação. Já a técnica de espectrofotometria por UV atende a princípios da química analítica verde, uma vez que evita a produção de resíduos em grande quantidade, dado que a utilização de reagente é mínima, o processo de preparo da amostra é simples, utiliza reagentes renováveis, não utiliza reagente tóxico, como o iodo, presente na titulação iodimétrica, mostra economia de tempo do analista e energia elétrica, além de apontar para a segurança do operador e dos resultados pela validação bem-sucedida18 , 38 , 41 .
CONCLUSÕES
As análises físico-químicas realizadas com os medicamentos referência, genéricos e similares da DIP, neste estudo, apontaram para a presença de não conformidades que sugerem impactos na efetividade do tratamento medicamentoso, como os valores de teor e de performance encontrados abaixo do preconizado pela FB 6 para o medicamento S2. O estudo também demonstrou que o teste farmacopeico por titulometria, para o doseamento da DIP, pode ser de difícil reprodução e que é possível quantificar o fármaco em comprimidos utilizando a espectrofotometria na região do UV, o que sugere que métodos farmacopeicos podem ser revistos em prol do alcance de resultados analíticos mais confiáveis.
Além disso, o monitoramento pós-comercialização de medicamentos com elevado padrão de consumo pela população, como é o caso, realizado por meio de avaliação da qualidade, pode sinalizar questões a serem investigadas pela vigilância sanitária, com o objetivo de apoiar sua missão de proteção à saúde da população.
Agradecimentos
Os autores agradecem a Universidade Federal da Bahia no nome da Pró-Reitoria de Ações Afirmativas e Assistência Estudantil (PROAE/UFBA); da Pró-Reitoria de Pesquisa, Criação e Inovação (PROPCI/UFBA) e da Pró-Reitoria de Ensino de Pós-Graduação (PROPG/UFBA).
REFERÊNCIAS
1. Kumar V, Abbas A, Fausto N, Aster JC. Robbins & Cotran patologia: bases patológicas das doenças. Rio de Janeiro: Elsevier; 2010[acesso 25 ago 2020]. Disponível em: https://www.academia.edu/36494092/_ROBBINS_and_COTRAN_2010_Bases_Patológicas_das_Doenças_8ed
2. Knappmann AL, Melo EB. Qualidade de medicamentos isentos de prescrição: um estudo com marcas de dipirona comercializadas em uma drogaria de Cascavel (PR, Brasil). Cienc Saude Coletiva. 2010;15(Supl.3):3467-76. https://doi.org/10.1590/S1413-81232010000900021
3. Jasiecka A, Ma¨slanka T, Jaroszewski JJ. Pharmacological characteristics of metamizole. Pol J Vet Sci. 2014;17(1):207-14. https://doi.org/10.2478/pjvs-2014-0030
4. Fundação de Vigilância em Saúde do Amazonas. Plano de contingência estadual para a infecção humana pelo SARS-CoV-2 (COVID-19). Manaus: Secretaria de Estado da Saúde; 2020[acesso 25 ago 2020]. Disponível em: https://www.saude.gov.br/images/pdf/2020/marco/11/PLANO-CORONAVIRUS-Amazonas.pdf
5. Conselho Regional de Farmácia de São Paulo – CRF-SP. Manual de orientação ao farmacêutico: COVID-19. São Paulo: Conselho Regional de Farmácia de São Paulo; 2020.
6. Fundação Oswaldo Cruz – Fiocruz. Plano de contingência da Fiocruz para pandemia de COVID-19 versão 1,3. Brasília: Fundação Oswaldo Cruz; 2020[acesso 25 ago 2020]. Disponível em: https://portal.fiocruz.br/documento/plano-de-contingencia-da-fiocruz-para-pandemia-de-covid-19-versao-13
7. Ministério da Saúde (BR). Relação nacional de medicamentos essenciais 2020. Brasília: Ministério da Saúde; 2020[acesso 25 ago 2020]. Disponível em: http://portalms.saude.gov.br/assistencia-farmaceutica/medicamentos-rename
8. Costa TMF, Lopes JC. Controle de qualidade de soluções orais de dipirona sódica 500 mg/mL referência, genérico e similar comercializadas em Gurupi TO. Amaz Sci Health. 2016;4(4):3-11. https://doi.org/10.18606/2318-1419/amazonia.sci.health.v4n4p3-11
9. Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC No31, de 11 de agosto de 2010. Dispõe sobre a realização dos estudos de equivalência farmacêutica e de perfil de dissolução comparativo. Diário Oficial União. 12 ago 2010.
10. Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC No37, de 3 de agosto de 2011. Dispõe sobre o guia para isenção e substituição de estudos de biodisponibilidade relativa/bioequivalência e dá outras providências. Diário Oficial União. 4 ago 2011.
11. Agência Nacional de Vigilância Sanitária – Anvisa. Instrução normativa No10, de 29 de setembro de 2016. Determina a publicação da lista de fármacos candidatos à bioisenção baseada no sistema de classificação biofarmacêutica (SCB) e dá outras providências. Diário Oficial União. 30 set 2016.
12. Agência Nacional de Vigilância Sanitária – Anvisa. Suspensos 124 lotes de medicamentos da Brainfarma. Notícias. 12 dez 2018[acesso 25 ago 2020]. Disponível em: http://portal.anvisa.gov.br/noticias/-/asset_publisher/FXrpx9qY7FbU/content/brainfarma-declara-recolhimento-de-124-produtos/219201/pop_up?inheritRedirect=false
13. Redação. Anvisa suspende venda de lote de dipirona sódica. Época Vida. 2 jul 2014[acesso 25 ago 2020]. Disponível em: https://epoca.globo.com/vida/noticia/2014/07/anvisa-suspende-venda-de-lote-de-bdipirona-sodicab.html
14. Lombardo M, Eserian JK. A análise da qualidade de medicamentos e o papel do laboratório oficial no contexto da saúde pública. Rev Adm Saude. 2017;17(67). https://doi.org/10.23973/ras.67.28
15. Rocha ACC, Silva ER, Braga RR. Controle de qualidade físico-químico de comprimidos de cloridrato de propanolol dispensados pelo programa farmácia popular do Brasil. Perspect Ciencia Tec. 2015;7(1):46-54.
16. Agência Nacional de Vigilância Sanitária – Anvisa. Farmacopeia brasileira. 6a ed. Brasília: Agência Nacional de Vigilância Sanitária; 2019[acesso 25 ago 2020]. Disponível em: http://portal.anvisa.gov.br
17. Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC No166, de 24 de julho de 2017. Dispõe sobre a validação de métodos analíticos e dá outras providências. Diário Oficial União. 25 jul 2017.
18. Cazedey ECL. Análise químico-farmacêutica e estudos de estabilidade e de dissolução de comprimidos de orbifloxacino [doutorado]. Araraquara: Universidade Estadual Paulista; 2012.
19. Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123-33. https://doi.org/10.1016/s0928-0987(01)00095-1
20. Khan KA. The concept of dissolution efficiency. J Pharm Pharmacol. 1975;27(1):48-9. https://doi.org/10.1111/j.2042-7158.1975.tb09378.x
21. Karageorgou E, Samanidou V. Youden test application in robustness assays during method validation. J Chromatogr A. 2014;1353:131-9. https://doi.org/10.1016/j.chroma.2014.01.050
22. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use – ICH. ICH harmonised tripartite guideline: validation of analytical procedures: text and methodology q2(r1). Geneva: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2005.
23. Instituto Nacional de Metrologia, Qualidade e Tecnologia – Inmetro. Orientação sobre validação de métodos analíticos: documento de caráter orientativo DOC-CGCRE-008. Brasília: Instituto Nacional de Metrologia, Qualidade e Tecnologia; 2020.
24. Lanna EG, Leão GR, Siqueira RA, Soares AF. Avaliação comparativa da qualidade de comprimidos de dipirona sódica referência, genérico e similar. Rev Cient Faminas. 2016;9(3):11-22.
25. Farias JF, Pelegrini DD. Avaliação comparativa da qualidade de comprimidos de dipirona similar em relação ao de referência. SaBios Rev Saude Biol. 2016;11(11):48-57.
26. Allen Jr LV, Popovich NG, Ansel HC. Formas farrnacêuticas e sistemas de liberação de fármacos. 9a ed. Porto Alegre: Artmed; 2013.
27. Órfão MK, Alves JO, Valladão AS, Bonfilio R, Andrighetti CR, Ribeiro EB et al. Controle de qualidade de comprimidos de hidroclorotiazida comercializados no município de Sinop MT. Sci Electron Arch. 2017;10(5):124-32.
28. Köhler F, Nascimento DH, Schwengber LL, Bandeira MP, Pazin V, Machado RP. Avaliação biofarmacotécnica e perfil de dissolução de comprimidos de dipirona: equivalência farmacêutica entre medicamentos de referência, genéricos e similares. Rev Bras Farm. 2009;90(4):309-15.
29. Midas Pharma. Metamizol midas: metamizol stada: mimetanal 500 mg tabletten, solution. Ingelheim: Midas Pharma; 2017.
30. Rao KVR, Devi KP, Buri P. Cellulose matrices for zero-order release of soluble drugs. Drug Dev Ind Pharm. 1988;14(15/17):2299-320. https://doi.org/10.3109/03639048809152017
31. Baveja SK, Rao KVR, Devi KP. Zero-order release hydrophilic matrix tablets of β-adrenergic blockers. Int J Pharm. 1987;39(1/2):39-45. https://doi.org/10.1016/0378-5173(87)90196-7
32. Schwartz JB, Simonelli AP, Higuchi WI. Drug release from wax matrices I: analysis of data with first‐order kinetics and with the diffusion‐controlled model. J Pharm Sci. 1968;57(2):274-7. https://doi.org/10.1002/jps.2600570206
33. Ofoefule SI, Udeogaranya PO, Okonta JM. Prediction of in vivo bioavailability of six brands of ciprofloxacin film coated tablets using the concept dissolution efficiency (DE). Boll Chim Farm. 2001;140(3):187-91.
34. Serra CHR, Storpirtis S. Comparação de perfis de dissolução da cefalexina através de estudos de cinética e eficiência de dissolução (ED%). Rev Bras Cienc Farm. 2007;43(1):79-88. https://doi.org/10.1590/S1516-93322007000100010
35. Skoug JW, Halstead G, Theis DL, Freeman JE, Fagan D, Rohrs B. Strategy for the development and validation of dissolution tests for solid oral dosage forms. Pharm Tech. 1996;20:58-72.
36. Manadas R, Pina ME, Veiga F. A dissolução in vitro na previsão da absorção oral de fármacos em formas farmacêuticas de liberação modificada. Rev Bras Cienc Farm. 2002;38(4):375-99. https://doi.org/10.1590/S1516-93322002000400002
37. Fernandes C, Mariano M, Naves R, Freitas T, Pinto M, Souza S et al. Controle de qualidade físico-químico de comprimidos de dipirona sódica adquiridos em drogarias de São Luis de Montes Belos (GO). Rev Eletr Fac Montes Belos. 2015;8(2):1-10.
38. Pereira PAP, Andrade JB. Fontes, reatividade e quantificação de metanol e etanol na atmosfera. Quim Nova. 1998;21(6):744-54. https://doi.org/10.1590/S0100-40421998000600015
39. Ayres GH. Evaluation of accuracy in photometric analysis. Anal Chem. 1949;21(6):652-7. https://doi.org/10.1021/ac60030a002
40. Association of Official Analytical Chemists – AOAC. Appendix f: guidelines for standard method performance requirements. Rockville: Association of Official Analytical Chemists; 2016[acesso 25 ago 2020]. Disponível em: http://www.eoma.aoac.org/app_f.pdf
41. Lenardão EJ, Freitag RA, Dabdoub MJ, Batista ACF, Silveira CC. Green chemistry: os 12 princípios da química verde e sua inserção nas atividades de ensino e pesquisa. Quim Nova. 2003;26(1):123-9. https://doi.org/10.1590/S0100-40422003000100020
Autor notes
Silva ES, Cardoso AST, Cazedey ECL – Concepção, planejamento (desenho do estudo), aquisição, análise, interpretação dos dados e redação do trabalho. Todas as autoras aprovaram a versão final do trabalho.
* E-mail: edith.cazedey@ufba.br
Declaração de interesses
Os autores informam não haver qualquer potencial conflito de interesse com pares e instituições, políticos ou financeiros deste estudo.
