ARTIGO
Recepção: 10 Março 2023
Aprovação: 15 Junho 2023
DOI: https://doi.org/10.22239/2317-269X.02161
RESUMO
Introdução: O novo marco regulatório de insumos farmacêuticos ativos compreende três resoluções editadas pela Agência Nacional de Vigilância Sanitária em 2020, RDC nº 359, RDC nº 361 e RDC nº 672, inaugurando novas abordagens para regularização do insumo farmacêutico ativo no país.
Objetivo: Demonstrar como a internalização dos requisitos legais e sanitários instituídos pelo novo marco regulatório de insumos ativos foi absorvida e implementada por Farmanguinhos, um laboratório farmacêutico oficial.
Método: Estudo transversal descritivo com base no levantamento de dados do arcabouço legal editado pela Agência Nacional de Vigilância Sanitária e a experiência prática de Farmanguinhos, o principal laboratório público fornecedor de medicamentos ao Ministério da Saúde.
Resultados: O principal resultado deste estudo foi o levantamento das necessidades para implementação do novo marco pelos laboratórios farmacêuticos oficiais, observando o maior rigor sanitário e regulatório imposto aos fabricantes de insumos ativos e a reflexão deste na proposta de adequação dos procedimentos operacionais de Farmanguinhos.
Conclusões: Cabe aos laboratórios farmacêuticos oficiais intermediar e atuar como facilitadores nas relações entre fabricantes de insumo farmacêutico ativo e autoridade sanitária. Assim, eles exercem um papel de impulsionador no estreitamento da relação com os fabricantes de insumos, por meio da revisão de seus procedimentos e edição de instrumentos de apoio, conduzindo a incorporação das exigências e em paralelo facilitando a otimização das atividades e ações voltadas à implementação da nova realidade regulatória, tanto internamente quanto pelos fabricantes de insumos.
Palavras chave: Insumo Farmacêutico Ativo, agência Nacional de Vigilância Sanitária, laboratório Farmacêutico Oficial, marco Regulatório.
ABSTRACT
Introduction: The new regulatory framework for active pharmaceutical ingredients comprises three resolutions edited by the National Health Surveillance Agency, the RDC No. 359, of March 27, 2020, the RDC No. 361, of April 1, 2020, and the RDC No. 672, of March 30, 2022.These regulations start new approaches to regularize the active pharmaceutical ingredient in the country.
Objective: To demonstrate how the internalization of the legal and health requirements established by the new regulatory framework for active ingredients was absorbed and implemented by Farmanguinhos, an Official Pharmaceutical Laboratory.
Method: Descriptive cross-sectional study based on data collection from the legal framework edited by the Brazilian Health Regulatory Agency, and the practical experience of Farmanguinhos, the main medicines supplier to the Ministry of Health.
Results: The main result of this study was the survey of the needs for the implementation of the new framework by the Official Pharmaceutical Laboratories, observing the greater sanitary and regulatory rigor imposed on manufacturers of active ingredients and the reflection of this in the proposed adequacy of operational procedures at Farmanguinhos.
Conclusions: This study concludes that it is up to the Official Pharmaceutical Laboratories to intermediate and act as facilitators in the relations between active pharmaceutical ingredients manufacturers, by reviewing their procedures and editing support tools, leading to the incorporation of requirements and, in parallel, facilitating the optimization of activities and actions aimed at the implementation of a new regulatory reality, both internally and by the manufacturers of inputs.
Keywords: Active Pharmaceutical Ingredient, brazilian Health Regulatory Agency, official Pharmaceutical Laboratory, regulatory Framework.
INTRODUÇÃO
Antes da criação da Agência Nacional de Vigilância Sanitária (Anvisa) em 1999, o sistema de vigilância sanitária não contemplava as informações e a documentação técnica a respeito dos insumos farmacêuticos ativos (IFA) e seus fabricantes. A partir do aprimoramento do arcabouço regulatório iniciado pela Anvisa já em 1999, as empresas farmacêuticas detentoras de registro no país ou com intenção de operar no país foram impulsionadas a passar por um processo interno de adequação, de forma a alcançar a capacitação necessária para submissão de um processo de registro ou de adequação de registro, em conformidade com os novos requisitos da legislação 1 .
Nesta trajetória de aprimoramento, é introduzida a exigência de estudos de equivalência farmacêutica e biodisponibilidade relativa e, em paralelo, a exigência de validação dos métodos analíticos para o produto acabado, assim como o maior controle sobre o fármaco e as informações técnicas oriundas do fabricante do fármaco 1 .
Desponta expressivamente com o início da Anvisa, a introdução e o crescente aumento de exigências técnicas sobre as informações a respeito do IFA advindas de seus fabricantes, como pode ser observado pelas Resoluções da Diretoria Colegiada (RDC) nº 133 2 , nº 134 3 , nº 135 4 e nº 136 5 , de 29 de maio de 2003, as quais estão intrinsecamente associadas à importância da qualificação destes fornecedores.
Com o aprimoramento do arcabouço regulatório da Anvisa, destaca-se a publicação da RDC nº 57, de 17 de novembro de 2009 6 , que dispôs sobre o registro de IFA no Brasil, no entanto, com abrangência limitada a uma lista restrita de IFA publicada pela Anvisa, por meio de instruções normativas (IN) distintas, as IN nº 15, de 17 de novembro de 2009 7 , e IN nº 3, de 28 de junho de 2013 8 , limitando a obrigatoriedade de registro sanitário de IFA a um número reduzido de 20 IFA, ou seja, dentro de um universo de medicamentos registrados em suas diversas classes terapêuticas, apenas 20 teriam seus IFA devidamente registrados e aprovados pela Anvisa, na forma de um dossiê submetido para esta finalidade.
Em 2014, ocorreu a consolidação das legislações sobre o registro de medicamentos novos, genéricos e similares em um único regulamento, a RDC nº 60, de 10 de outubro de 2014, e consequentemente a equiparação dos requisitos técnicos para os medicamentos e para os IFA 9 .
A RDC nº 60/2014 também traz um incremento da documentação técnica a ser apreciada pela Anvisa, acrescentando o relatório de desenvolvimento do produto, assim como o estudo da compatibilidade do IFA com os excipientes 9 .
Esta resolução sofreu atualizações em 2017, 2020 e em 2022, com a edição da RDC nº 200, de 29 de janeiro de 2017, da RDC n° 361, de 1º de abril de 2020, e a RDC n° 753, de 28 de setembro de 2022, respectivamente, sendo o ano de 2020 marcado pela introdução do novo marco regulatório (MR) na legislação brasileira de registro 10 , 11 , 12 .
Em 2022, a RDC n° 200/2017 foi substituída pela RDC n° 753/2022, no entanto, não houve atualizações no texto e nas determinações já trazidas pelo novo MR de IFA, assim como não ocorreram alterações nas diretrizes voltadas aos requisitos da tecnologia farmacêutica. As atualizações inauguradas por este regulamento reposicionam os requisitos de segurança e eficácia em um novo patamar regulatório, uma vez que determina duas novas vias de registro distintas conceitualmente para os medicamentos novos e inovadores, ao mesmo tempo em que amplia os caminhos elegíveis à comprovação de segurança e de eficácia 12 .
A evolução das exigências regulamentares para o IFA tem, em sua linha do tempo, total coerência com o caminho percorrido pela Anvisa focado na convergência pelas melhores práticas regulatórias internacionais, culminando com sua atuação como membro regulador do International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), tal como resumido pela Figura 1 , com a trajetória da legislação de medicamentos no que se refere à inserção e à evolução das legislações próprias sobre IFA.
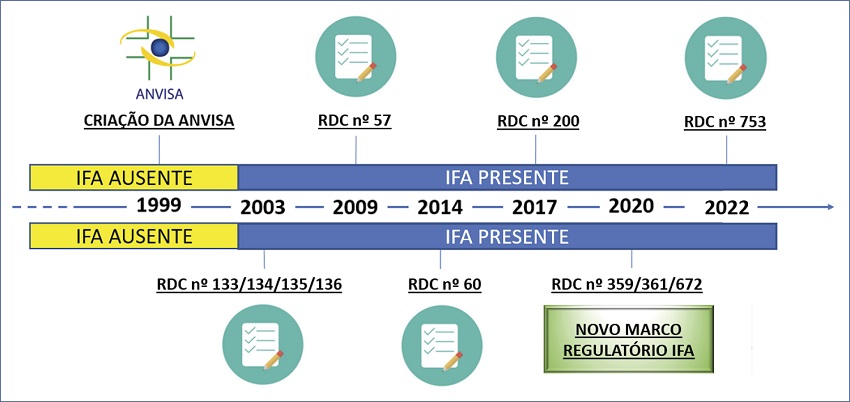
Figura 1
Linha do tempo das legislações que compõem o arcabouço legal com a presença do insumo farmacêutico ativo (IFA).
Fonte: Elaborada pelos autores, 2023.
Em 2020, o novo MR de IFA compreendeu três resoluções editadas pela Anvisa simultaneamente, as RDC nº 359, de 27 de março 12 , RDC nº 361 11 , e a RDC nº 362, de 27 de março, revogada pela atual RDC nº 672, de março de 2022 14 , inaugurando novas abordagens para regularização do IFA no país. Um dos regulamentos estabelece a instituição do dossiê do insumo farmacêutico ativo (Difa) e da carta de adequação do dossiê do insumo farmacêutico ativo (Cadifa), aplicáveis a todos os IFA que integram a formulação de medicamentos no Brasil 13 .
A regulamentação do novo MR é o resultado da internalização dos guias do comitê gestor do ICH 13 .
Uma nova prática foi inaugurada pela RDC nº 359/2020, permitindo a empresas sem Cadastro Nacional da Pessoa Jurídica (CNPJ) no país e sediadas no exterior a realizarem o processo de peticionamento da Cadifa e do cadastro de empresa diretamente junto à Anvisa 13 .
Desponta-se uma nova perspectiva regulatória, sendo o fabricante do IFA responsável por responder diretamente à Anvisa sobre seus processos, sem a intermediação da indústria farmacêutica.
Ressalta-se que a sistematização de aprovação do Difa e da Cadifa, assim como a certificação de boas práticas de fabricação abrangem todo IFA a ser empregado em medicamentos objeto de registro na Anvisa, ou seja, todos os fabricantes de IFA, nacionais ou internacionais, que possuem seus insumos na composição de medicamentos regulados pela Anvisa, estão sujeitos às disposições do novo MR 13 .
As exigências regulatórias destinam-se aos setores regulados público e privado, na mesma medida. A regulamentação não discrimina IFA para doenças negligenciadas e seus fabricantes, ou seja, não há excepcionalidades. Desta forma, os laboratórios farmacêuticos oficiais (LFO) são diretamente impactados no âmbito do compromisso de atendimento com medicamentos aos diversos Programas de Saúde Pública do Ministério da Saúde (MS), como tuberculose e malária e, por conseguinte, pelo acesso da população brasileira a estes tratamentos.
Este cenário no campo das doenças negligenciadas se deve ao fato dos LFO serem os principais e muitas vezes os únicos fornecedores destes medicamentos ao MS, por não serem produtos atrativos ao setor privado. De mesmo modo, a indústria internacional de IFA não conta com muitas opções e empresas interessadas em atendimento regulatório rigoroso e fornecimento com este patamar de exigências.
Este estudo foi realizado visando demonstrar como a internalização dos requisitos legais e sanitários instituídos pelo novo MR de IFA editado pela Anvisa pode ser absorvida por um LFO, por meio da apresentação da experiência de implementação deste marco por Farmanguinhos, com foco no suporte da relação deste com os fabricantes de IFA e sua intermediação junto às autoridades de saúde, Anvisa e MS, contribuindo na manutenção do abastecimento de medicamentos no país.
Desta forma, o trabalho propôs modificações em procedimentos dos LFO aplicáveis ao IFA e seus fabricantes, perpassando a seleção e qualificação de fornecedores, o registro de medicamentos e o ciclo de vida do produto.
MÉTODO
Neste estudo, a primeira etapa consistiu na análise descritiva com o levantamento de dados do arcabouço regulatório sanitário, compreendido pelas RDC, IN, guias e perguntas e respostas, referentes ao registro sanitário de medicamentos novos, genéricos e similares, assim como aqueles relacionados diretamente ao tema IFA.
Os dados foram coletados por meio do sítio eletrônico oficial da Anvisa em https://www.gov.br, utilizando a busca da biblioteca temática de medicamentos, insumos farmacêuticos e a busca por meio do consolidado do estoque regulatório, da própria base de dados da Anvisa. O levantamento da legislação ocorreu em outubro de 2022, observando o período de publicações de 1999, referente ao ano de criação da Anvisa, até 2022.
Para a análise da legislação sanitária de registro de medicamentos anterior à criação da Anvisa, foram utilizados os dados dos dossiês de registro submetidos anteriormente à criação da Anvisa e aprovados pelo MS, com pesquisa na base de dados interna do LFO em estudo.
A segunda etapa deste estudo consistiu na experiência prática de Farmanguinhos, um LFO vinculado ao MS do Brasil. Foi realizada em uma avaliação dos procedimentos vigentes no ano de 2022 e identificados os procedimentos que apresentavam ações e atividades vinculadas ou a IFA ou a fabricantes de IFA.
Após análise dos procedimentos selecionados, estes foram confrontados com os novos requisitos técnico-regulatórios estabelecidos pelas legislações que compõem o novo MR de IFA, vigente em 2022.
Com base neste estudo e em reuniões multidisciplinares sobre a implementação do novo marco, foram propostas adequações nestes procedimentos, visando incorporar os requerimentos do novo marco nestas atividades da indústria farmacêutica. Na proposta de adequação foram considerados os seguintes cenários relatados nas reuniões multidisciplinares:
-
O novo MR afeta distintamente os fabricantes de IFA nacionais e internacionais, visto que, em termos de regulamentação os fabricantes nacionais e internacionais de IFA são impactados de forma distinta;
-
A avaliação dos problemas relatados pelos fabricantes de IFA para doença negligenciada para fornecimento destes IFA e como sua qualificação à luz da legislação brasileira tem se tornado um processo mais rigoroso no âmbito sanitário.
RESULTADOS E DISCUSSÃO
O novo MR de IFA exige dos seus fabricantes um rigor maior na composição do dossiê e na interação direta com o órgão regulador brasileiro. Esta nova legislação pode ter reflexo nos programas de saúde pública, uma vez que, segundo Chaves et al. 15 , o Sistema Único de Saúde (SUS) tem atuação direta na assistência farmacêutica integral voltada à oferta de medicamentos pelo setor público, responsável pela promoção do acesso universal, que, em muitos casos, são comprados exclusivamente pelo setor público, uma característica específica de aquisição no mercado brasileiro. Consequentemente, esta compra pode ser impactada pela queda na oferta de IFA por empresas estrangeiras.
A cadeia produtiva de medicamentos na indústria farmacêutica é iniciada pelo IFA, desta forma um problema de qualidade ou de fornecimento afeta toda a cadeia produtiva de medicamentos. Como nos Estados Unidos, aqui no Brasil, a principal causa de desabastecimento é motivada pela falta de insumos, seguido de outros motivos. Nosso país se encontra muito vulnerável a este cenário, devido ao exponencial aumento de sua dependência de importação de insumos farmacêuticos 16 .
Em contrapartida, o novo MR constituiu um avanço regulatório e sanitário na regulamentação de IFA e seus fabricantes, por ampliar sua aplicabilidade a todos os insumos integrantes da formulação de medicamentos registrados ou submetidos a registro no país. A implantação do MR a todos os IFA atende uma demanda antiga do setor nacional de fármacos, reprimindo a entrada de IFA oriundo do exterior, sem a chancela da Anvisa para as boas práticas de fabricação, a preços inferiores a indústria nacional 17 .
As farmoquímicas nacionais são empresas já reguladas pela Anvisa e formam um parque industrial fabril no Brasil composto por 49 empresas ativas, concentradas majoritariamente na Região Sul-Sudeste do país. Desde a publicação da RDC nº 69, de 8 de dezembro de 2014, que dispôs sobre as boas práticas de fabricação de insumos, adotando o guia internacional ICH Q7, que trata do mesmo tema, a Anvisa iniciou em 2015 sua participação nas inspeções das farmoquímicas paralelamente à implementação de novos procedimentos em nível tripartite, com o objetivo de harmonizar as ações locais das vigilâncias sanitárias 18 .
Segundo levantamento realizado por Pinto et al. 19 , o não cumprimento das boas práticas de fabricação é majoritariamente o principal motivo de recolhimento de IFA entre 2011 e 2019. Com o novo marco este cenário mudará na medida em que os fabricantes internacionais de IFA serão submetidos à avaliação da Anvisa e somente os fabricantes aptos permanecerão no mercado, mitigando os problemas de recolhimento. A qualificação de fornecedores será apoiada pela Anvisa e seus procedimentos serão amparados pelo novo MR de IFA.
As fabricantes de IFA nacionais na condição de empresas reguladas pela Anvisa já têm interface com as diversas áreas técnicas da agência, o que tem se intensificado com as novas práticas regulatórias inauguradas pelo novo marco e permitiu que elas assumam a dianteira de uma vantagem regulatória em relação às empresas farmoquímicas internacionais e, em paralelo, apresentem potencial para se tornarem mais competitivas do ponto de vista técnico para atuação em mercados mais regulados.
Por outro lado, os LFO que têm o compromisso de sustentar as políticas de saúde nacionais, especialmente pelo desenvolvimento e produção de medicamentos para doenças negligenciadas, os quais não são atrativos para a indústria privada 20 , se deparam com o desafio adicional de manter a adesão destes fabricantes de IFA em face da maior regulação sanitária sobre os IFA e seus fabricantes, por meio de suporte técnico na implantação do novo marco e uma programação de demanda para aquisições destes IFA, repercutindo em uma parceria entre LFO e fabricante de IFA.
De acordo com a Organização Mundial da Saúde, as doenças negligenciadas são aquelas que atingem populações vulneráveis e apresentam diversas lacunas de diagnóstico e tratamento, delegando à população um fardo econômico e social devastador 21 .
Neste caso específico, os procedimentos de seleção de novos fornecedores e o procedimento de qualificação destes com a introdução de critérios técnicos à luz do novo marco são essenciais na mitigação de risco de aprovação de fabricantes de IFA internacionais que não estarão aptos a cumprir com os requisitos e serem excluídos após um extenso trabalho já realizado. No campo dos IFA para doenças negligenciadas, poderá haver o estreitamento das relações entre Anvisa, MS, LFO e fabricante de IFA e, aos LFO, caberá atuar como intermediadores tanto entre os fabricantes de IFA e Anvisa, facilitando as ações regulatórias determinadas pelo novo MR, quanto entre o MS e Anvisa, sendo antecipadores no caso de identificação de problemas no atendimento ao novo marco com possível risco de desabastecimento.
A Associação Brasileira das Indústrias de Química Fina, Biotecnologia e suas Especialidades tem destacado que um país sem fabricação própria de IFA está vulnerável às intempéries do cenário internacional e não se alavanca sem a promoção de uma política pública consistente que favoreça a produção local, dando sustentabilidade ao sistema de saúde 22 .
Historicamente, houve um vertiginoso crescimento na dependência do Brasil aos insumos oriundos de empresas no exterior. Desde a década de 1990, houve uma deterioração da competitividade da indústria nacional, alavancada pelo processo de abertura comercial 23 . Desta forma, a aplicabilidade majoritária do novo marco de IFA recai para os fabricantes de fármaco internacionais, dentre eles destaca-se a China como o principal fornecedor de IFA e intermediários 24 .
Assim, permanecemos dependentes dos fabricantes internacionais, com agravamento deste cenário, pela possibilidade de não adesão dos fabricantes destes IFA no exterior aos rigorosos critérios do novo MR, representados pelo alto padrão das exigências regulamentares do ICH, incorporadas em documentos, estudos e evidências técnicas exigidos pelo novo MR, sendo sua fidelização ao MR brasileiro de extrema importância, uma vez que são os principais fornecedores dos produtos regularizados no país.
Não seria o momento de impulsionar as farmoquímicas nacionais? Um caminho seria a oferta de incentivos a estas empresas, visando impulsionar seu portfólio de IFA, com prerrogativas às empresas que trabalhem com IFA para doenças negligenciadas, diminuindo a dependência destes insumos provenientes do exterior e o risco de a desistência destas empresas atuarem no Brasil devido às exigências regulamentares da Anvisa.
Atividades de um laboratório farmacêutico oficial impactadas pelo novo marco regulatório
Após o levantamento do arcabouço regulatório até 2022, foi realizado o levantamento dos procedimentos operacionais padrão de Farmanguinhos que possuem atividades relacionadas ou afetadas pelos requisitos do novo MR.
Os procedimentos diretamente afetados por novas atividades inerentes à introdução do novo MR foram relacionados a: processo de seleção de candidatos a fornecedores; qualificação de fornecedores de IFA; requisitos de qualidade e regulatórios que devem ser cumpridos durante a vigência do registro do medicamento, estabelecidos nos documentos designados na indústria farmacêutica como acordo de qualidade e controle de mudanças; registro e pós-registro; processo de compra e produção do medicamento.
O novo MR alterou a dinâmica de interação entre o fabricante do IFA, o fabricante do medicamento e o órgão regulador no Brasil, assim como estabeleceu um novo canal administrativo diretamente com o fabricante do IFA, no que tange a análise de sua documentação, eximindo a indústria farmacêutica desta responsabilidade no dossiê do medicamento. Por outro lado, o novo marco delega à indústria farmacêutica maior necessidade de monitoramento e controle sistemático sobre o fabricante do fármaco, assumindo o papel-chave no controle do ciclo de vida do IFA, o qual refletirá no controle empregado na sua aquisição, sua liberação e uso na produção e na expedição dos medicamentos fabricados pelos LFO.
Com a vigência do novo MR, o fluxo da preparação do dossiê de registro e/ou alteração de registro sofreu mudanças, tornando-se estratégico o programa de auditorias presenciais por equipes do LFO, permitindo o estreitamento da parceria, de forma a otimizar o alcance dos alinhamentos e compreensão aos critérios de qualidade exigidos pela Anvisa, constituindo-se uma relação imprescindível à mitigação dos riscos de não cumprimento aos impactos regulatórios decorrentes do ciclo de vida do IFA.
A adequada implementação do novo MR pelos LFO, assim como sua atuação junto aos fabricantes de fármacos, visando a sua internalização e cumprimento, terá grande impacto no papel dos 21 LFO, distribuídos por todo o território nacional e vinculados aos governos estaduais e federal, uma vez que atendem ao SUS com 30% dos medicamentos 25 , preenchendo lacunas de fornecimento no campo das doenças infecciosas e negligenciadas e diminuindo a dependência do país 26 .
Considerando que o IFA representa em muitos casos 70% a 80% do preço final do medicamento e que no Brasil 23 , ainda que os LFO tenham como característica a dependência de insumos a partir de indústrias nacionais ou estrangeiras 27 , não sendo historicamente produtores de matérias-primas, é evidente que seu papel é imprescindível tanto pela vertente da formulação quanto do custo deste medicamento, sendo possível admitir que problemas de aquisição com o IFA provenientes de problemas técnicos ou de custo terão impacto direto na oferta e acesso a tais medicamentos.
A compilação do formato documental do Difa para o Commom Technical Document (CTD) padronizou a estrutura do Difa de diferentes procedências. No entanto, a similaridade documental dos fabricantes não é suficiente, uma vez que três parâmetros adicionais são importantes, como disponibilidade do IFA, seu custo e qualidade 28 .
Os LFO se deparam facilmente com problemas de oferta de fabricantes de IFA para doenças negligenciadas capazes de conciliar estes parâmetros e por esta razão, ao prestarem todo o suporte e monitoramento do processo de implantação deste marco pelos fabricantes dos fármacos, principalmente os internacionais, podem propiciar, a partir de então, que estes estabeleçam uma nova forma de atuação com a autoridade sanitária brasileira, a Anvisa, balizados pelas principais normativas internacionais na área dos insumos farmacêuticos.
Os laboratórios privados e públicos são distintamente afetados, na medida em que IFA de alto custo e qualidade são mais facilmente adequados ao novo marco, ao passo que IFA de baixo custo e volume de compra, para doença negligenciada, apresentam chances de não alcançar os requerimentos de qualidade, diante da resistência de seus fabricantes em se adequar ao novo marco. Um exemplo é evidenciado pela experiência de recusa destes em realizar estudos de estabilidade em zona climática IVb, exigida para fornecimento ao Brasil 29 .
O LFO poderá, em alguns casos, prestar um suporte ativo, como o caso do estudo de estabilidade, e se propor a realizar o estudo, de forma a garantir a manutenção do fornecimento para o Brasil e mitigar o risco de desabastecimento.
Os LFO, na condição de laboratórios públicos, devem obedecer aos procedimentos específicos de aquisição de insumos em geral por meio de processos licitatórios.
Não há uma segmentação da legislação sanitária para LFO, no entanto, diversas atividades de um LFO são regidas por meio de um arcabouço legal específico, a exemplo do sistema de aquisição de IFA e sua compra, regido pela Lei de Pregão, a Lei n° 14.133, de 1° de abril de 2021, que estabelece normas gerais de licitação e contratação para a administração pública direta. Este processo se torna mais distinto ainda ao se conciliar a necessidade de compra pelo menor preço e cumprimento de requisitos técnicos, visando atendimento à Anvisa e ao registro sanitário do medicamento.
Desta forma, é proposto que, dentro do procedimento de seleção de empresas candidatas a fornecedores de IFA para o LFO, seja inserido o Termo de Compromisso, configurando um pré-requisito ao prosseguimento das etapas subsequentes.
O Termo de Compromisso dará ciência aos candidatos sobre os novos termos sobre a relação do fabricante do IFA com a autoridade sanitária do Brasil e com o próprio LFO e remeterá novas exigências técnicas à luz das disposições do novo MR, delimitando a participação para as próximas etapas de somente candidatos dispostos e cientes de seus compromissos a partir das exigências trazidas pela RDC nº 359/2020, que institui o Difa e a Cadifa.
A verificação preliminar pode compreender um questionário ao fabricante do IFA contendo perguntas objetivas pretendidas ao entendimento do grau de conhecimento e adesão destes laboratórios ao novo MR.
O Quadro 1 apresenta um modelo de questionário com perguntas direcionadas à área técnica/qualidade do fabricante do IFA, com o objetivo de mapear o estágio de conformidade ou viabilidade de conformidade que estas empresas apresentam. Tal questionário não visa ser um documento de cunho excludente, mas um subsídio para uma análise de risco aprofundada na qualificação deste fornecedor.
Modelo de questionário exploratório: verificação do status do fabricante de insumos farmacêuticos ativos perante novo marco regulatório.

IFA: Insumo farmacêutico ativo; Difa: Dossiê do insumo farmacêutico ativo; Cadifa: Carta de adequação de dossiê de insumo farmacêutico ativo; CPBF: Certificado de boas práticas de fabricação; PIC: Pharmaceutical Inspection Cooperation Scheme ; RDC: Resolução da Diretoria Colegiada.
Somado aos subsídios fornecidos pelo questionário como suporte à avaliação de risco, esse pode retratar o estágio de conformidade ao novo MR que determinado fabricante de fármaco se encontra, assim como a viabilidade ou não de sua adequação aos critérios impostos por este marco.
Procedimento de qualificação de fornecedores
O procedimento operacional de qualificação de fornecedores de IFA deve prever os novos requisitos delegados aos fabricantes de fármacos com a vigência do novo MR de IFA e esta análise juntamente com o processo de certificação da Anvisa elevam o rigor regulatório na qualificação destes fornecedores.
Tais procedimentos aplicados nas diversas etapas que compõem todo o processo de qualificação de fornecedores de IFA são essenciais para a segurança da seleção, qualificação e certificação de empresas fabricantes de fármaco aderentes às determinações impostas pela legislação sanitária brasileira, assim como garantia de manutenção do atendimento deste fornecedor aos requisitos sanitários, sem prejuízo do fornecimento dos referidos IFA, indispensáveis à fabricação dos medicamentos.
A Figura 2 sintetiza as principais etapas a serem percorridas na qualificação de fornecedor dentro do contexto de atuação de um laboratório farmacêutico.
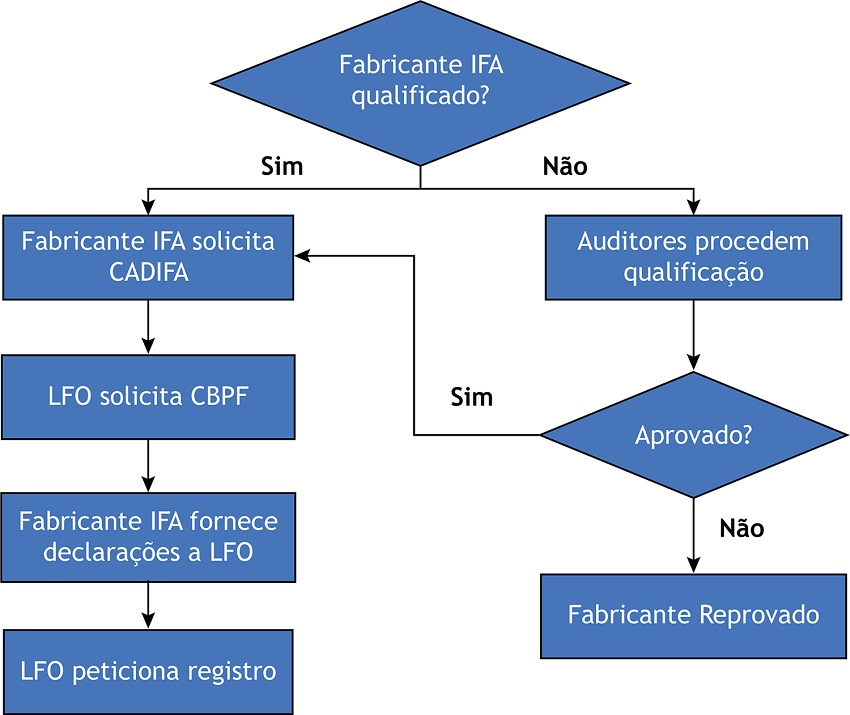
Figura 2
Principais etapas na qualificação de fornecedor de insumos ativos após marco regulatório.
Fonte: Elaborada pelos autores, 2023.IFA: Insumo farmacêutico ativo; Cadifa: Carta de adequação de dossiê de insumo farmacêutico ativo; CBPF: Certificado de boas práticas de fabricação; LFO: Laboratório farmacêutico oficial.
Análise de risco na qualificação de fornecedores
O gerenciamento de riscos configura uma das práticas mais recentes internalizadas nos requerimentos regulatórios internacionais relacionados à qualidade de medicamentos. A análise de risco é uma ferramenta importante para detectar o nível de capacidade de adesão do futuro fornecedor ao novo marco e, se pertinente, a exclusão deste nesta etapa.
A RDC n° 672/2022 estabelece condições para o cumprimento das boas práticas de fabricação do IFA, as quais são contempladas pela autoridade sanitária do Brasil, considerando resultados das autoridades internacionais e auditorias pelas empresas fabricantes dos medicamentos, assim como as não conformidades relatadas em relatórios de inspeção e as suas ações corretivas realizadas
Adicionalmente, é estimulado que seja realizada a análise de risco e seja determinada uma escala de pontuação para o critério “Regularização do Fabricante de Fármaco na Anvisa”, atribuindo o escalonamento: alto, médio e baixo risco.
O Quadro 2 compila os critérios propostos para a análise de risco e sua respectiva classificação de risco dos fabricantes de IFA sujeitos ao processo de qualificação de fornecedor.
Critérios de risco à luz do novo marco regulatório dos insumos farmacêuticos ativos segundo RDC n° 359/2020 e RDC n° 361/2020.

*Módulo: conjunto de documentos que compõem partes do dossiê no formato CTD, como previsto na RDC n° 359/2020. Anvisa: Agência Nacional de Vigilância Sanitária; IFA: Insumo farmacêutico ativo; Difa: Dossiê de insumo farmacêutico ativo; Cadifa: Carta de adequação de dossiê de insumo farmacêutico ativo; CPBF: Certificado de boas práticas de fabricação; PIC: Pharmaceutical Inspection Cooperation Scheme ; RDC: Resolução da Diretoria Colegiada; CTD: Common Technical Document; ICH: International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use .
Auditorias de qualidade na certificação de fornecedores
As auditorias de qualidade in loco são essenciais na verificação da eficiência e eficácia do sistema de qualidade implementado e do cumprimento das boas práticas de fabricação, o que é amparado pelo próprio regulamento da Anvisa que instituiu as diretrizes gerais de boas práticas de fabricação, a RDC n° 658/2022 30 .
Dentro do escopo do programa de inspeção para estabelecimentos internacionais fabricantes de IFA instituído pela Anvisa, e ainda, por meio da instituição da Cadifa, torna-se estratégico o programa de auditorias presenciais por equipe de auditores do LFO, permitindo o estreitamento da parceria, de forma a otimizar o alcance dos alinhamentos e a compreensão dos critérios de qualidade exigidos pela Anvisa, constituindo-se uma relação imprescindível à mitigação dos riscos de não cumprimento aos impactos regulatórios decorrentes do ciclo de vida do IFA.
Previsão do novo marco regulatório nos acordos de qualidade
O fornecedor de IFA qualificado deve obedecer aos requisitos estabelecidos à manutenção de sua condição de certificação como fornecedor de IFA, como a observância às disposições do acordo de qualidade, principalmente no que se refere aos compromissos de comunicação das mudanças, suas notificações e submissões junto à Anvisa.
De forma a auxiliar estes processos de mudanças e seus devidos desdobramentos, atividades de controle e monitoramento podem ser utilizadas, como observado na Figura 3 .
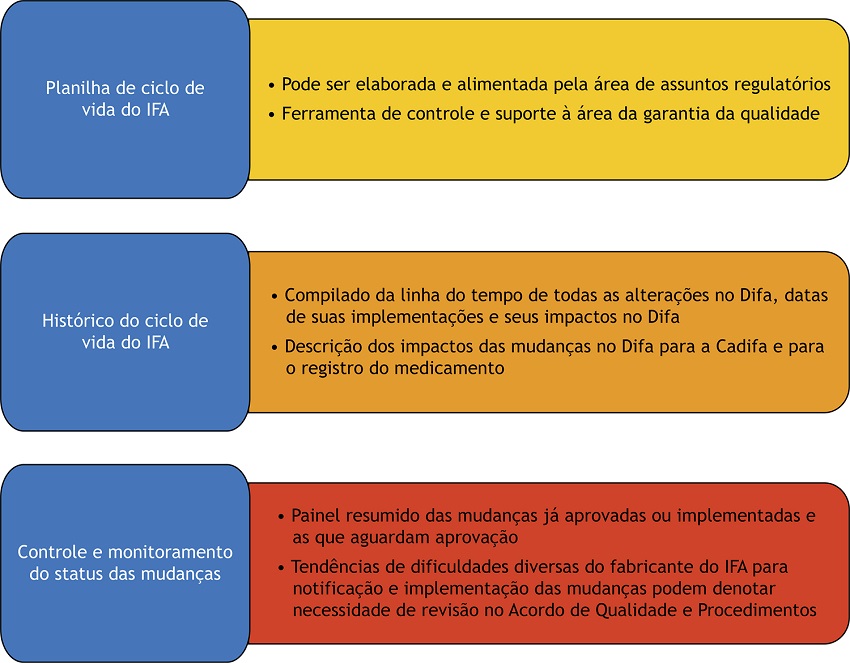
Figura 3
Atividades de controle e monitoramento dos processos de mudança.
Fonte: Elaborada pelos autores, 2023.IFA: Insumo farmacêutico ativo; Difa: Dossiê de insumo farmacêutico ativo; Cadifa: Carta de adequação de dossiê de insumo farmacêutico ativo.
Fluxo de adequação do fabricante do IFA e medicamento antecedente à petição de registro, no registro e pós-registro
Posteriormente ao processo de qualificação de fornecedor, tanto os fabricantes de IFA quanto os fabricantes de medicamentos estarão aptos a prosseguir com as seguintes etapas rumo a obtenção da Cadifa e registro sanitário de medicamento.
O fluxo demonstra as etapas antecedentes à petição de registro ou pós-registro demandadas com a vigência do novo MR:
-
LFO envia legislação/guia ao fabricante do IFA e, se for o caso, fornece treinamento adequado;
-
LFO fornece suporte técnico e faz a intermediação do processo de cadastro da empresa e da submissão da Cadifa pelo fabricante do IFA;
-
O fabricante do IFA solicita a Cadifa pelo sistema eletrônico de peticionamento da Anvisa designado sistema Solicita;
-
LFO submete a petição de Certificação Boas Práticas de Fabricação para o fabricante do IFA;
-
LFO protocola a petição de registro/pós-registro de medicamento relacionada ao Difa e seu respectivo fabricante.
Os processos de registro de medicamentos pertencentes às categorias de novos, inovadores, genéricos e similares devem ser instruídos de acordo com o checklist documental estabelecido pela RDC nº 753/2022.
A avaliação e a decisão sobre a petição pós-registro específica a ser submetida devem ser provenientes da comunicação dos fabricantes de IFA com Cadifa sempre que houver alterações no Difa. O detentor de registro do medicamento deve avaliar se houve revisão da Cadifa e protocolar as mudanças pós-registro correspondentes, conforme disposto na RDC nº 73/2016, com redação dada pela RDC nº 361/2020.
Como incorporar e registrar documentalmente os requisitos do ciclo de vida do dossiê do insumo farmacêutico ativo?
A documentação do controle de mudanças constitui-se uma ferramenta crucial no encadeamento correto das aprovações e implementações das mudanças por ambas as partes, fabricante de IFA e fabricante de medicamento.
Por meio do monitoramento, espera-se obter os dados das versões atualizadas dos documentos, para que haja a divulgação interna aos setores interessados e abastecimento de novos dados ao próprio sistema informatizado, se cabível, a respeito das versões aprovadas para uso a partir de uma data definida.
O conhecimento e o acesso às versões possuem o propósito de mitigar risco na aquisição e no uso de IFA em desacordo com as condições aprovadas na Cadifa e no registro sanitário do medicamento correspondente.
Preferencialmente, o sistema informatizado de gestão empresarial adotado pela empresa poderá atribuir um número de controle de versão do IFA aprovada passível de alteração, acoplada à sua codificação ou, se independente, vinculada a esta.
Monitoramento da implementação da mudança (fabricante do insumo farmacêutico ativo x fabricante do medicamento)
O LFO deve elaborar uma sistemática de monitoramento das mudanças realizadas no Difa e no registro do medicamento, assumindo o papel-chave no controle do ciclo de vida do IFA, o qual refletirá o controle empregado na aquisição de IFA, sua liberação e uso na produção e na expedição dos medicamentos fabricados pelos LFO, com garantia de conformidade do IFA utilizado na formulação ao Difa aprovado na Anvisa para este medicamento.
Tal monitoramento por parte dos LFO também permitirá um suporte técnico destes aos fabricantes do IFA, mitigando risco de infração sanitária, como o equívoco do fabricante do IFA exportar lotes de IFA não conformes à versão aprovada para o DIFA e para o medicamento em questão.
Controle de compra do insumo farmacêutico ativo de acordo com o novo marco regulatório para produção
As indústrias farmacêuticas possuem seus sistemas próprios de gestão administrativa. Os LFO por serem empresas públicas, utilizam o sistema eletrônico de informações (SEI) em seus processos de aquisição de IFA e demais insumos utilizados na fabricação dos medicamentos. Em particular, para os IFA adquiridos na rotina de produção dos medicamentos, é necessário constar na justificativa da compra as informações preliminares do fabricante de fármaco aprovado para este medicamento. Com a implantação do novo MR de IFA, é fundamental que dados adicionais sejam acrescentados para referenciar as características corretas do IFA aprovado e a Cadifa correspondente, assim como o código/referência do Difa em questão. Em paralelo, a planilha do ciclo de vida do IFA contendo as alterações aprovadas para o IFA dentro do processo do registro do medicamento serve de instrumento de suporte.
CONCLUSÕES
A evolução das exigências regulatórias para o IFA tem em sua linha do tempo total coerência com o caminho percorrido pela Anvisa focado na convergência pelas melhores práticas regulatórias internacionais, culminando com sua atuação como membro regulador do ICH.
É imprescindível que, em paralelo ao aprimoramento do arcabouço regulatório aplicável aos IFA e medicamentos que os utilizam, as políticas públicas voltadas aos programas de saúde pública para doenças negligenciadas observem este novo cenário regulatório, por um lado desafiador para os fabricantes de IFA nacionais e internacionais e para os fabricantes de medicamentos, mas que, por outro lado, eleva o nível de segurança e eficácia destes insumos e medicamentos, assim como torna as empresas fabricantes de IFA nacional mais competitivas no mercado internacional.
A amplificação do rigor sanitário aplicável a todos os fabricantes de insumos farmacêuticos e, por conseguinte, a todas as formulações de medicamentos trazidas pelo novo MR de insumos ativos, deve motivar o estreitamento das relações entre os LFO, os fabricantes de IFA, a Anvisa e o MS, visando a manutenção do fornecimento dos medicamentos com qualidade assegurada e continuidade do adequado abastecimento para atendimento às necessidades da população.
Conclui-se com os resultados deste estudo que cabe ao LFO intermediar e atuar como facilitador nas relações e atividades entre o LFO, os fabricantes de IFA e a autoridade sanitária regulamentadora do novo MR, tendo um papel de impulsionador no estreitamento da relação com os fabricantes de IFA, por meio da revisão de seus procedimentos operacionais e edição de instrumentos de apoio, como os manuais, os quais podem mitigar os principais entraves a sua absorção do novo MR, conduzindo a incorporação das exigências e em paralelo facilitando a otimização das atividades e ações voltadas à implementação da nova realidade regulatória, tanto internamente pelo LFO quanto pelos fabricantes de IFA.
REFERÊNCIAS
Costa SM. Regulamentação da Anvisa para registro de medicamentos: uma análise da evolução regulatória para medicamentos genéricos, similares e novos e seu impacto no contexto de um laboratório farmacêutico oficial [dissertação de mestrado]. Rio de Janeiro: Fundação Oswaldo Cruz; 2017.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 133, de 29 de maio de 2003. Dispõe sobre o registro de medicamento similar e dá outras providências. Diário Oficial União. 30 maio 2003.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 134, de 29 de maio de 2003. Dispõe sobre a adequação dos medicamentos já registrados. Diário Oficial União. 30 maio 2003.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 135, de 29 de maio de 2003. Aprova o regulamento técnico para medicamentos genéricos. Diário Oficial União. 30 maio 2003.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 136, de 29 de maio de 2003. Dispõe sobre o registro de medicamento novo. Diário Oficial União. 30 maio 2003.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 57, de 17 de novembro de 2009. Dispõe sobre o registro de insumos farmacêuticos ativos e dá outras providências. Diário Oficial União. 18 nov 2009.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 15, de 17 de novembro de 2009. Dispõe sobre prazos e cronograma do registro de insumos farmacêuticos ativos (IFA) e as priorizações para 1ª etapa de implantação do registro de IFA. Diário Oficial União. 18 nov 2009.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 3, de 28 de junho de 2013. Dispõe sobre prazos e cronograma do registro de insumos farmacêuticos ativos (IFA) e as priorizações para 2ª etapa de implantação do registro de IFA. Diário Oficial União. 29 jun 2013.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 60, de 10 de outubro de 2014. Dispõe sobre os critérios para a concessão e renovação de registro de medicamentos sintéticos e semissintéticos, classificados como novos, genéricos e similares, e dá outras providências. Diário Oficial União. 11 out 2014.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 200, de 26 de dezembro de 2017. Dispõe sobre os critérios para a concessão e renovação do registro de medicamentos com princípios ativos sintéticos e semissintéticos, classificados como novos, genéricos e similares, e dá outras providências. Diário Oficial União. 28 dez 2018.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 361, de 27 de março de 2020. Altera a RDC Nº 200, de 26 de dezembro de 2017 e a RDC Nº 73, de 7 de abril de 2016, para dispor sobre a submissão do dossiê de insumo farmacêutico ativo (DIFA) no registro e pós-registro de medicamento, respectivamente. Diário Oficial União. 1 abr 2020.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 359, de 27 de março de 2020. Institui o dossiê de insumo farmacêutico ativo (DIFA) e a carta de adequação de dossiê de insumo farmacêutico ativo (CADIFA). Diário Oficial União. 1 abr 2020.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 753, de 28 de setembro de 2022. Dispõe sobre o registro de medicamentos de uso humano com princípios ativos sintéticos e semissintéticos, classificados como novos, inovadores, genéricos e similares. Diário Oficial União. 5 out 2022.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 672, de 30 de março de 2022. Dispõe sobre os critérios de boas práticas de fabricação e institui o programa de inspeção para estabelecimentos internacionais fabricantes de insumos farmacêuticos ativos. Diário Oficial União. 31 mar 2022.
Chaves CC, Hasenclever L, Oliveira MA. Redução de preço de medicamento em situação de monopólio no Sistema Único de Saúde: o caso do tenofovir. Physis. 2018;28(1):1-26. https://doi.org/10.1590/S0103-73312018280103
Oliveira AG, Silveira D. Políticas públicas: reavaliar para tornar o Brasil um país capaz de suprir suas necessidades na produção de IFA. Infarma. 2022;34(2):107-9. https://doi.org/10.14450/2318-9312.v34.e2.a2022.pp107-109
Arrepia DB, Costa JCS, Tabak D. Registro de insumos farmacêuticos ativos: impactos e reflexos sobre as indústrias farmoquímicas e farmacêutica instaladas no Brasil. Vigil Sanit Debate. 2015;3(2):9-19. https://doi.org/10.3395/2317-269x.00439
Agência Nacional de Vigilância Sanitária – Anvisa. Revisão anual das inspeções em farmoquímicas nacionais: Coins (RINIFA_2020). Brasília: Agência Nacional de Vigilância Sanitária; 2021[acesso 7 jan 2023]. Disponível em: https://www.gov.br/anvisa/pt-br/centraisdeconteudo/publicacoes/fiscalizacao-e-monitoramento/fiscalizacao/relatorio-de-revisao-anual-das-inspecoes-farmoquimicas-nacionais-coins-2020
Pinto NN, Resende KA, Couto RO. Insumos farmacêuticos ativos irregulares no Brasil: análise descritiva de 2011 a 2019. Vigil Sanit Debate. 2021;9(1):61-70. https://doi.org/10.22239/2317-269X.01456
Figueiredo TA, Neto RG, Magalhães JL. A produção pública de medicamentos no Brasil. Rev Saúde Colet. 2021;26(Supl.2):3423-34. https://doi.org/10.1590/1413-81232021269.2.04572020
World Health Organization – WHO. Ending the neglect to attain the sustainable development goals: a road map for neglect tropical diseases 2021-2030. Geneva: World Health Organization; 2020[acesso 10 dez 2022]. Disponível em: https://www.who.int/neglected_diseases/revised-draft-NTD-Roadmap23apro2020.pdf
Associação Brasileira das Indústrias de Química Fina, Biotecnologia e suas Especialidades – ABIFINA. O desafio da fabricação local de IFA. Rio de Janeiro: Associação Brasileira das Indústrias de Química Fina, Biotecnologia e suas Especialidades; 2021[acesso 6 ago 2022]. Disponível em: https://abifina.org.br
Dias ECF, Ambrosino MCP, Oliveira NR, Magalhães JL. A dependência de insumos farmacêuticos importados no Brasil: um estudo de caso do medicamento antirretroviral nevirapina no laboratório farmacêutico oficial farmanguinhos. Rev Gest Sist Saúde. 2016;5(2):125-34. https://doi.org/10.5585/rgss.v5i2.194
Cherian JJ, Rahi M, Singh S, Reddy SE, Gupta YK, Katoch VM et al. India’s road to independence in manufacturing active pharmaceutical ingredients: focus on essential medicines. Economies. 2021;9(2):1-18. https://doi.org/10.3390/economies9020071
Ministério da Saúde (BR). Assistência farmacêutica: laboratórios oficiais. Brasília: Ministério da Saúde; 2022[acesso 4 dez 2022]. Disponível em: http://www.saude.gov.br/assistencia-farmaceutica/laboratorios-oficiais
Silva SM, Figueiredo TA, Magalhães JL. Proposta para o alinhamento entre o plano estratégico institucional de um laboratório farmacêutico oficial e o departamento de TI. Rev Inov Proj Tec. 2020;8(2):198-220. https://doi.org/10.5585/iptec.v8i2.18660
Fernandes DR, Gadelha CA, Maldonado JM. Vulnerabilidades das indústrias nacionais de medicamentos e produtos biotecnológicos no contexto da pandemia de COVID-19. Cad Saúde Pública. 2021;37(4):1-14. https://doi.org/10.1590/0102-311X00254720
Alan Z, Kaur S, Porwal PK. Understanding the problems in pharmaceutical in procurement with special reference to active pharmaceutical ingredients and excipients. Accred Qual Assur. 2018;23:319-28. https://doi.org/10.1007/s00769-018-1344-6
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 318, de 6 de novembro de 2019. Estabelece os critérios para a realização de estudos de estabilidade de insumos ativos e medicamentos, exceto biológicos. Diário Oficial União. 7 nov 2019.
Agência Nacional de Vigilância Sanitária – Anvisa. Resolução RDC N° 658, de 30 de março de 2022. Dispõe sobre as diretrizes gerais de boas práticas de fabricação de medicamentos. Diário Oficial União. 31 mar 2022.
Autor notes
* E-mail: soraya.costa@fiocruz.br
Declaração de interesses
Os autores informam não haver qualquer potencial conflito de interesse com pares e instituições, políticos ou financeiros deste estudo.
