Article / Clinical Case Report
Primary splenic angiosarcoma: a rare entity often associated with rupture and hemoperitoneum
 Jean Michel Correia Monteiro
Mirella Nardo
Fernando Peixoto Ferraz de Campos
Jean Michel Correia Monteiro
Mirella Nardo
Fernando Peixoto Ferraz de Campos
Primary splenic angiosarcoma: a rare entity often associated with rupture and hemoperitoneum
Autopsy and Case Reports, vol. 9, no. 3, e2019100, 2019
São Paulo, SP: Universidade de São Paulo, Hospital Universitário
Received: 07 May 2019
Accepted: 06 June 2019
DOI: 10.4322/acr.2019.100
Abstract: Primary splenic angiosarcoma (PSA) is a rare neoplasm of vascular origin associated with aggressive behavior and poor prognosis. The clinical presentation is usually non-specific and is mostly characterized by a wasting disease with anemia and splenomegaly, mimicking a wide range of entities. The authors present the case of an 80-year-old woman with cardiovascular comorbidities with a 6-month history of weight loss, fatigue, weakness, pallor, and abdominal pain. The physical examination showed massive splenomegaly and pallor. After a thorough evaluation that ruled out lymphoproliferative diseases, the working diagnosis was a myelodysplastic disorder. A few days after discharge, she returned to the emergency room with severe abdominal pain, worsening fatigue, and a remarkable pallor. Point-of-care ultrasound showed free intraperitoneal fluid. Spleen rupture was confirmed by abdominal computed tomography (CT) scan, and an emergency laparotomy with splenectomy was performed. The postoperative period was uneventful, and the patient recovered in a few days. The histopathology confirmed the diagnosis of PSA and the patient was referred to an oncological center. Two months later staging CT demonstrated liver and peritoneal metastases, and despite the chemotherapy she died 6 months after the diagnosis.
Keywords: Splenic Diseases, Splenomegaly, Hemangiosarcoma, Splenic Rupture.
INTRODUCTION
Primary splenic angiosarcoma (PSA) is a malignant mesenchymal-derived elongated endothelial cell originating from the splenic sinusoidal endothelial cells. 1 PSA is a rapidly proliferating, highly infiltrating, and widely spreading neoplasm, despite its insidious, often asymptomatic, presentation. 2 Clinical manifestations are non-specific, including fatigue, abdominal pain, anorexia, weight loss, and anemia. Similarly, the imaging studies are often non-specific; however, splenomegaly with enlargement greater than 12 cm length, in the presence of a complex mass and hypervascular tumors may be found. 3 The diagnosis is challenging and is often made only after splenectomy. More than 300 PSA cases have been described. 4 Azevedo et al. 5 reported the case of a 57-year-old woman previously treated for breast ductal carcinoma, and as far as we know, to date, our case was the first case report of PSA with spontaneous rupture in a patient in Brazil.
Our report is to remind physicians to include this entity among the differential diagnoses for splenomegaly. We also would like to highlight the first reported case of PSA with spontaneous rupture in Brazil, based on a literature review in the PubMed, Scielo, Lilacs, Web of Science databases, over an unlimited period, using the keyword “Primary Splenic Angiosarcoma
CASE REPORT
An 80-year-old woman was admitted with a 6-month history of progressive abdominal pain, weight loss, fatigue, and pallor. Her past medical history included hypertension and atherosclerotic cardiovascular disease. The physical evaluation was generally unremarkable except for massive splenomegaly and pallor. The spleen edge was palpable 10 cm below the left costal margin, firm, and painless. There was no palpable lymphadenopathy. The laboratory work-up showed anemia (hemoglobin was 8.9 g/dL; reference range [RR]: 12.3–15.3 g/dL) with mild microcytosis (MCV = 79.3 fL/red cell) and 1.36% of reticulocytes. The white blood cell and platelet counts were within the reference range and distribution. Renal and liver function test results were in the reference range with a ferritin level of 206 ng/mL (RR: 13–150 ng/mL) and transferrin saturation of 10% (RR: 20%–50%). The folic acid and vitamin B12 determinations were unremarkable. An abdominal computed tomography (CT) scan revealed a 24 × 11 × 18 cm diffusely heterogeneous spleen with hypovascular areas with a volume of 2470 mL (RR: 107.2–314.5 mL). The liver was unremarkable, and no evidence of venous thrombosis or abdominal lymphadenopathy was found ( Figure 1).
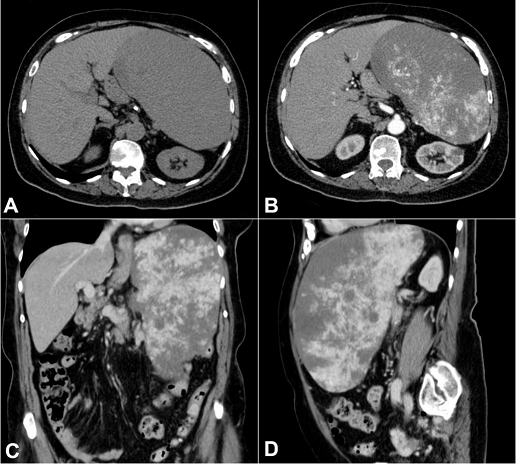
Figure 1
Abdominal CT. A and B – axial plane; C – coronal plane; D – sagittal plane; B, C and D – after contrast enhancement showing large, expansive, and hyper vascularized mass within the spleen with wash-out, displacing adjacent structures. No lymphadenomegaly was observed.
Chest CT scan suggested cardiomegaly and coronary atherosclerotic disease. Upper digestive endoscopic evaluation revealed a small gastric polyp, which was revealed to be a foveolar polyp. A bone marrow biopsy disclosed an increased population of the three cellular lineages for the patient’s age; approximately 60% cellularity with increased granulocytic precursors, suggestive of retarded maturation, including 1% of CD34 immature cells. There was no morphological evidence of lymphoma and no reticulin fibrosis. The patient was discharged and referred to an outpatient clinic follow-up with the working diagnosis of splenomegaly secondary to myelodysplastic syndrome. One week later, the patient returned to the emergency room complaining of acutely worsening abdominal pain. There was no history of trauma before the onset of symptoms. Physical examination showed hemodynamic instability (mean arterial pressure of 57 mmHg, pulse rate of 86 bpm), with marked pallor, and diffuse abdominal tenderness. Laboratory studies revealed a hemoglobin value of 5.7 g/dL with mild microcytosis (MCV = 79.1 fL/red cell) and leukocytosis. The platelet count and renal and liver functions remained in reference ranges. Serologic studies for HIV, hepatitis B, and C, and syphilis were negative. After clinical stabilization and packed red blood cell transfusion, a point-of-care ultrasound revealed free intraperitoneal fluid. The abdominal CT scan confirmed mild hemoperitoneum attributable to spleen rupture and an emergency laparotomy with splenectomy was performed. The postoperative period was uneventful, and the patient was discharged after 6 days of hospitalization. Two months after discharge, the patient was re-evaluated and the CT images showed metastases in the liver and peritoneum.
PATHOLOGICAL REPORT
The spleen was enlarged, weighing 1200g (RR: 112g) and measuring 18.0 × 13.0 × 6.5 cm. The splenic capsule had multiple disrupted areas with fibrin deposition. Macroscopic sections showed multiple ill-defined nodular areas; the larger ones were white and firm and smaller ones were blue-red and spongy ( Figure 2). On microscopy, the spleen parenchyma was replaced by anastomosing, irregularly ectatic vascular channels lined by plump mildly atypical lining cells. Intercalated solid fusiform areas consisted of spindle cell fascicles showing moderate nuclear atypia, with frequent mitotic figures. These closely packed vascular channels were filled with red blood cells. Small eosinophilic globules identified in the cytoplasm of the spindle cells were highlighted by periodic-acid Schiff (PAS) reagent ( Figures 3 and 4).
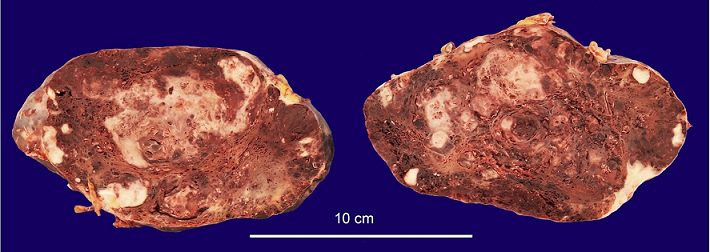
Figure 2
Gross view of the spleen, showing ill-defined, white nodular areas with firm consistency intercalated with smaller blue-red, spongy nodules.
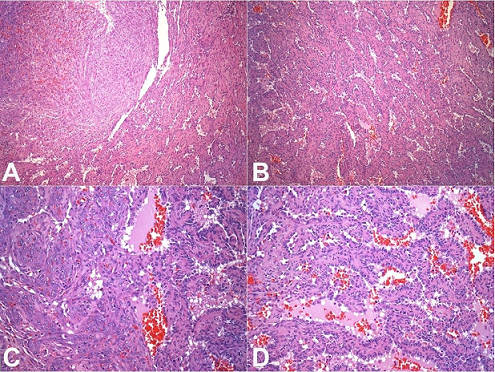
Figure 3
Photomicrographs of the spleen. A – Solid area showing spindle cells fascicles representing closed packed vascular spaces intercalated with anastomosing vascular channels (H&E, 100X); B – Spongy area showing irregular anastomosing vascular channels lined by plump cells (H&E, 100X); C – Transition between a solid area with spindle cells showing mild to moderate nuclear atypia and a spongy area (H&E, 200X); D – Irregular anastomosing vascular channels lined by plump cells with mild nuclear atypia (H&E, 200X).
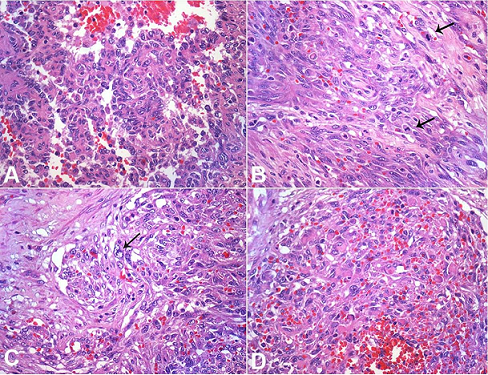
Figure 4
Photomicrographs of the tumor highlighting the atypical features. A – Plump cells with mild nuclear atypia lining the spongy areas. Note the intermediate nuclear sized and irregular nuclear contour (H&E, 400X); B – Fusocellular areas showing frequent mitosis (arrows; H&E, 400X); C – Enlarged and irregular nuclei in the spindle cell areas (arrow; H&E, 400X); D – Presence of small eosinophilic globules in the cytoplasm of spindle cells (H&E, 400X).
Immunohistochemistry demonstrated diffuse positivity for CD31 and FLI-1 in both the spongy and the solid fusiform areas. However, CD34 was reactive only in the fusiform areas. D2-40 was not demonstrated. There was no positive expression for CD8, CD68, lysozyme or CD21 on the tumor cells. The Ki-67 index was increased (about 50%) in the spindle cell areas when compared to the spongy areas (about 10%) ( Figures 5 and 6). Despite the morphological aspects of littoral cell hemangioma, the presence of atypia, the high index of the Ki67 and the immunophenotype were consistent with angiosarcoma.
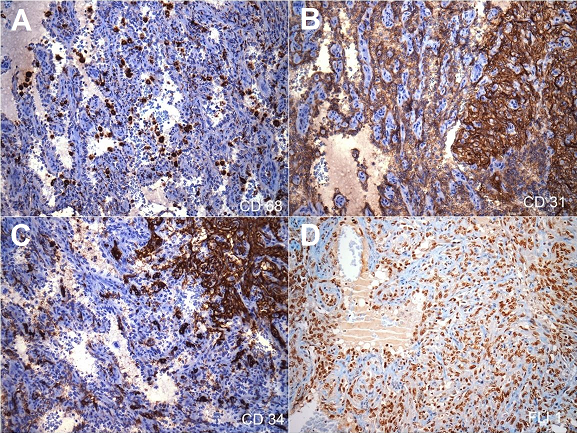
Figure 5
Photomicrographs of the spleen (all pictures 200X) – Immunohistochemical panel. A – CD68 was negative in the cells lining the vascular channels. Note the presence of histiocytes internal positive control; B – Strong and diffuse CD31 positivity in both spindle cells and plump cells lining the anastomosing channels; C – CD34 was positive in the solid area with spindle cells; D – Strong and diffuse FLI-1 positivity in both spindle cells and plump cells lining the anastomosing channels.
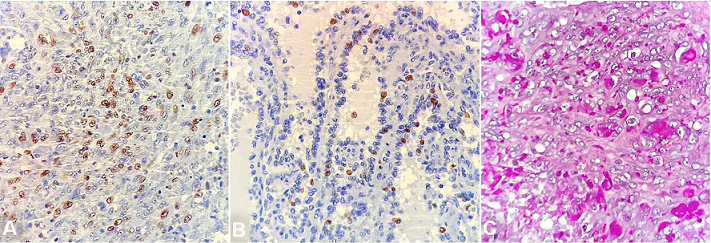
Figure 6
Photomicrographs of immunohistochemistry marker Ki-67 showing the higher index in the spindle cell area (50%) than the spongy area (10%) ( A and B – 400X). PAS staining highlights the eosinophilic intracytoplasmic globules, as well as some histiocytes ( C – 600X).
DISCUSSION
Originally described by Theodor Langhans 6 in 1879, the PSA is one of the rarest human malignancies, with an estimated annual incidence of 0.14 to 0.25 per million people. PSA is the rarest type of angiosarcoma and accounts for 2.6% or less of all soft tissue angiosarcomas. 2,7-9 Notwithstanding, PSA is the most frequent non-lymphoid primary neoplasm of the spleen. 10 PSA is thought to arise from the splenic red pulp’s immature endothelial-type cells. 11 This entity usually is highly aggressive with a metastatic rate ranging from 69% to 100%; metastases are mostly seen in the liver, lungs, lymph nodes, and bones. 1,12 The dismal prognosis may reflect the indolent presentation with a relatively late-stage diagnosis. Benign splenic tumors, such as hemangiomas or hemangioendotheliomas, have been suggested as precursors to PSA. 7,12,13 PSA has been reported in association with environmental or occupational exposure to carcinogens as ionizing radiation (e.g. thorium dioxide), arsenic, vinyl chloride, and chemotherapy for lymphoma, these causal relationships have not been unequivocally substantiated, and most cases occur without identifiable risk factors. 4,14,15 PSA is generally a neoplasm of middle-aged and older people with the mean age at presentation of 50–60 years, but cases in young people and children also have been reported. 7,16-19 A slight male predilection has been observed. Most clinical manifestations are variable and unspecific, such as fatigue, anorexia, weight loss, and abdominal pain. The laboratory work-up shows anemia, and eventually leukopenia or leukocytosis (more common), and thrombocytopenia. Serum lactate dehydrogenase (LDH) value is often elevated, and consumption coagulopathy has been reported. 12,20 Splenomegaly is present in 85% of cases and may be the single manifestation. 1,12 In up to 30% of cases, the first presentation consists of an acute abdomen with uncontrolled hemorrhagic shock due to spontaneous splenic rupture. 7,12
Imaging diagnosis of PSA remains challenging because of the overlap with other vascular splenic tumors. The imaging features vary according to the examination technique modality; however, splenomegaly is virtually always evident. Ultrasound depicts a heterogeneous echotexture complex solid mass with an area of necrosis, and hemorrhage. Cystic formations within the mass are occasionally seen with associated increased vascularity on Doppler examination. On the CT, solitary or multiple nodular masses that exhibit heterogeneous contrast enhancement are present. Necrosis and hemorrhagic foci likely contribute to the heterogeneity of the images, and hemoperitoneum is generally recognized in cases of spontaneous rupture. No particular calcification pattern is described. The magnetic resonance imaging shows areas of increased and decreased signal intensity in both T1- and T2-weighed sequences, reflecting a solid tumor with areas of necrosis. 12,21,22
Pathologic findings include splenomegaly with a mean splenic weight of 1073g (RR: 112g). 12 The tumor descriptions range from firm and well-circumscribed to poorly-limited spongy tumor, with hemorrhagic, necrotic, or nodular lesions, and cystic spaces. Scattered tumor areas within the spleen can be seen but eventually the neoplastic process replaces the entire splenic parenchyma. 6,12 Our patient is an elderly woman with clinical and radiological features consistent with the reported characteristics of PSA, and no history of risk factor exposure was identified. Most often PSA is both nodular with relatively homogeneous features associated with poorly circumscribed infiltrative tumor. A vasoformative component consistent with disorganized anastomosing vascular channels lined by atypical endothelial cells with bizarre giant cells are often seen. These giant cells were not found in our case. In addition, intracytoplasmic hyaline globules can be identified with H&E staining; the globules are highlighted with the PAS reaction. Eosinophilic nuclear bodies in the atypical cells are sometimes present. Vascular spaces may present as “slit-like,” “honeycomb-like,” or capillary-like patterns. A solid sarcomatous pattern interfaces with the vasoformative regions. Sheets of malignant epithelioid cells with round to irregular nuclei with vesicular nuclear chromatin may be present. The histological appearance and grade do not directly correlate with the outcome, although the mitotic index and the tumor size are considered prognostic factors. 18 Hemophagocytosis, hemosiderin, extramedullary hematopoiesis, amyloid, and calcification may be found in variable degrees within the tumoral mass. 12 The immunohistochemical profile is characterized by the presence of endothelial markers such as CD31, CD34, FVIII-RA, VEGFR3, and FLI 1. At least two of these markers are required for the diagnosis. Although the presence of histiocytic markers has been considered a clue for the diagnosis of PSA in some reports, they are not ubiquitous. Neuhauser et al. 12 reported that 89% of cases (24/27) exhibited positivity to one or both histiocytic markers (56% were positive for CD68, and 56% for lysozyme). Therefore, “the lack of histiocytic markers may not dissuade from diagnosis of angiosarcoma, if atypia and increased mitotic activity, shared with angiosarcomas, are present” (Dr. Neuhauser personal communication). The histomorphological features of the index case are those of PSA, confirmed by the immunohistochemical markers of vascular origin. The presence of atypia and the high Ki67 index, plus the absence of histiocytic markers in tumor cells helped rule out the diagnosis of littoral cell angioma, which is the main differential. In the series of Falk et al. 7; only 1 case in 23 presented positivity for CD68. These markers are not found in angiosarcoma of sites other than the spleen. The mitotic index by Ki 67 usually varies between 5% and 10%. In the case reported by Coppola et al., 15 the positivity for CD 68 was interpreted as possible littoral cell origin of the neoplastic cells. Littoral cell angiomas are splenic vascular tumors that mimic the findings of hypovascular angiosarcomas but generally do not diffusely involve the spleen and do not show atypia. The differential diagnosis of splenomegaly with a complex mass includes hemangiomas, littoral cell angioma, lymphangioma, hemangiopericytoma, lymphoid-origin neoplasia, and metastatic disease. In the setting of metastatic disease, ovarian carcinoma is among the more common primary tumors that metastasizes to the spleen and should be considered in the differential diagnosis although the histologic features are not those of PSA. 23 The presence of concomitant malignant tumors in patients with PSA—markedly breast cancer, lymphoma, colon cancer, skin cancer, and renal cell carcinoma—has been reported. 24
In a review 4 of 110 Chinese PSA cases, nearly 70% had metastases by the time of diagnosis, and 60% presented as splenic rupture. Moreover, some series reported a rate of 100% of metastasis to liver, bone, and bone marrow, as well as a poor prognosis with a survival rate of 20% in 6 months. 3,4,7,12,14-17 Splenic biopsy is generally contraindicated because of the high risk of inducing rupture and may not render the diagnosis. Reliable diagnosis is made after splenectomy and histological study, 25 although diagnosis with fine-needle aspiration cytology has been reported. 26
The treatment of PSA is challenging. 14 Splenectomy is the main treatment for PSA and ideally should be performed before splenic rupture, with direct impact on the survival time. Even though there is no standard chemotherapy regimen, epirubicin and ifosfamide, 7 paclitaxel, 27 and the same chemotherapy regimen as used for follicular lymphoma 28 have been reported as chemotherapy options. There is one case of high-dose chemotherapy followed by auto-PBSCT (peripheral blood stem cell transplantation), in which the patient had 5 years of progression-free survival, but eventually died after recurrence of disease. 29 Other drugs are bevacizumab 30 and immunotherapy with rIL-2. 31,32 The first one, which is a recombinant humanized antibody against the vascular endothelial growth factor, has been used in some case reports for angiosarcoma from other sites.
At the time of our patient’s splenectomy, the abdominal cavity’ did not show evidence of metastases, and neither did the imaging exams. However, in keeping with the known aggressive behavior of PSA, she presented liver and peritoneal metastases two months after surgery. Clinical deterioration (Karnofsky Performance Status 70% and PS-ECOG 2), and daily febrile reactions with no evidence of infection, as well as evidence of PSA at other sites, 33 prompted treatment with paclitaxel. We also started non-selective β-AR antagonists because of her cardiovascular disease, as we considered that it may have a role in controlling angiosarcoma, based on previous data. 34 However, our patient died of hepatic failure due to massive hepatic infiltration and sepsis. Although no metastases were found at the time of diagnosis and on the laparotomy, she died in 6 months after the diagnosis.
REFERENCES
Xu L, Zhang Y, Zhao H, Chen Q, Ma W, Li L. Well-differentiated angiosarcoma of spleen: a teaching case mimicking hemangioma and cytogenetic analysis with array comparative genomic hybridization. World J Surg Oncol. 2015;13(1):300. http://dx.doi.org/10.1186/s12957-015-0716-1. PMid:26462621.
Hamid KS, Rodriguez JA, Lairmore TC. Primary splenic angiosarcoma. JSLS. 2010;14(3):431-5. http://dx.doi.org/10.4293/108680810X12924466006521. PMid:21333203.
Thompson WM, Levy AD, Aguilera NS, Gorospe L, Abbott RM. Angiosarcoma of the spleen: imaging characteristics in 12 patients. Radiology. 2005;235(1):106-15. http://dx.doi.org/10.1148/radiol.2351040308. PMid:15749977.
Li R, Li M, Zhang LF, et al. Clinical characteristics and prognostic factors of primary splenic angiosarcoma: a retrospective clinical analysis from China. Cell Physiol Biochem. 2018;49(5):1959-69. http://dx.doi.org/10.1159/000493656. PMid:30235449.
Azevedo OS, Santos BN, Liboni NS, Costa JF, Campos OD. Splenic angiosarcoma: a diagnostic splenectomy finding. Case Rep Oncol. 2016;9(3):733-7. http://dx.doi.org/10.1159/000452619. PMid:27920710.
Langhans T. Pulsirende cavernöse Geschwulst der Milz mit metastatischen Knoten in der Leber. Tödtlicher Verlauf binnen 5 Monaten. Arch Pathol Anat Physiol Klin Med. 1879;75(2):273-91. http://dx.doi.org/10.1007/BF02134657.
Falk S, Krishnan J, Meis JM. Primary angiosarcoma of the spleen. A clinicopathologic study of 40 cases. Am J Surg Pathol. 1993;17(10):959-70. http://dx.doi.org/10.1097/00000478-199310000-00001. PMid:8372948.
Shukla M, Shukla VK, Basu S, Kumar M. Fever, anemia and splenomegaly: a rare presentation of splenic angiosarcoma. Indian J Med Paediatr Oncol. 2011;32(4):230-2. http://dx.doi.org/10.4103/0971-5851.95148. PMid:22563160.
Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. Lancet Oncol. 2010;11(10):983-91. http://dx.doi.org/10.1016/S1470-2045(10)70023-1. PMid:20537949.
Kohutek F, Badik L, Bystricky B. Primary angiosarcoma of the spleen: rare diagnosis with atypical clinical course. Case Rep Oncol Med. 2016;2016:4905726. http://dx.doi.org/10.1155/2016/4905726. PMid:27867672.
Takato H, Iwamoto H, Ikezu M, Kato N, Ikarashi T, Kaneko H. Splenic hemangiosarcoma with sinus endothelial differentiation. Acta Pathol Jpn. 1993;43(11):702-8. PMid:8310831.
Neuhauser TS, Derringer GA, Thompson LDR, et al. Splenic angiosarcoma: a clinicopathologic and immunophenotypic study of 28 cases. Mod Pathol. 2000;13(9):978-87. http://dx.doi.org/10.1038/modpathol.3880178. PMid:11007038.
Yang KF, Li Y, Wang DL, Yang JW, Wu SY, Xiao WD. Primary splenic angiosarcoma with liver metastasis: a case report and literature review. World J Gastroenterol. 2016;22(12):3506-10. http://dx.doi.org/10.3748/wjg.v22.i12.3506. PMid:27022233.
Deng R, Chang W, Wu X, Chen J, Tao K, Zhang P. Primary splenic angiosarcoma with fever and anemia: a case report and literature review. Int J Clin Exp Pathol. 2015;8(11):14040-4. PMid:26823717.
Coppola S, Leva A, Pagni F, Famularo S, Gianotti L. Demanding diagnosis of splenic angiosarcoma as cause of delayed treatment of spontaneous splenic rupture: a case report and literature review. Case Rep Surg. 2017;2017:6256102. http://dx.doi.org/10.1155/2017/6256102. PMid:28261515.
Serrano OK, Knapp E, Huang K, et al. Pediatric primary splenic angiosarcoma: an aggressive multidisciplinary approach to the oncologic management of a rare malignancy. World J Surg Oncol. 2014;12(1):379. http://dx.doi.org/10.1186/1477-7819-12-379. PMid:25487642.
Badiani R, Schaller G, Jain K, Swamy R, Gupta S. Angiosarcoma of the spleen presenting as spontaneous splenic rupture: a rare case report and review of the literature. Int J Surg Case Rep. 2013;4(9):765-7. http://dx.doi.org/10.1016/j.ijscr.2013.06.007. PMid:23856255.
Naka N, Ohsawa M, Tomita Y, et al. Prognostic factors in angiosarcoma: a multivariate analysis of 55 cases. J Surg Oncol. 1996;61(3):170-6. http://dx.doi.org/10.1002/(SICI)1096-9098(199603)61:3<170::AID-JSO2>3.0.CO;2-8. PMid:8637202.
Chen KT, Bolles JC, Gilbert EF. Angiosarcoma of the spleen: a report of two cases and review of the literature. Arch Pathol Lab Med. 1979;103(3):122-4. PMid:581838.
Aqil B, Green LK, Lai S. Primary splenic angiosarcoma associated with anemia, leukocytosis and thrombocytopenia. Ann Clin Lab Sci. 2014;44(2):217-21. PMid:24795063.
Abbott RM, Levy AD, Aguilera NS, Gorospe L, Thompson WM. From the archives of the AFIP: primary vascular neoplasms of the spleen: radiologic-pathologic correlation. Radiographics. 2004;24(4):1137-63. http://dx.doi.org/10.1148/rg.244045006. PMid:15256634.
Thompson WM, Levy AD, Aguilera NS, Gorospe L, Abbott RM. Angiosarcoma of the spleen: imaging characteristics in 12 patients. Radiology. 2005;235(1):106-15. http://dx.doi.org/10.1148/radiol.2351040308. PMid:15749977.
Valbuena JR, Levenback C, Mansfield P, Liu J. Angiosarcoma of the spleen clinically presenting as metastatic ovarian cancer. A case report and review of the literature. Ann Diagn Pathol. 2005;9(5):289-92. http://dx.doi.org/10.1016/j.anndiagpath.2005.03.007. PMid:16198958.
Hsu JT, Chen HM, Lin CY, et al. Primary angiosarcoma of the spleen. J Surg Oncol. 2005;92(4):312-6. http://dx.doi.org/10.1002/jso.20419. PMid:16299797.
Duan YF, Jiang Y, Wu CX, Zhu F. Spontaneous rupture of primary splenic angiosarcoma: a case report and literature review. World J Surg Oncol. 2013;11(1):53. http://dx.doi.org/10.1186/1477-7819-11-53. PMid:23497454.
Singh P, Sharma S, Gupta P, Lal A, Srinivasan R. Primary splenic angiosarcoma with liver metastasis: a rare neoplasm diagnosed on fine-needle aspiration cytology and cell block immunocytochemistry. J Cytol. 2018;35(2):114-6. http://dx.doi.org/10.4103/JOC.JOC_148_16. PMid:29643660.
Casper ES, Waltzman RJ, Schwartz GK, et al. Phase II trial of paclitaxel in patients with soft-tissue sarcoma. Cancer Invest. 1998;16(7):442-6. http://dx.doi.org/10.3109/07357909809011697. PMid:9774950.
Zwi LJ, Evans DJ, Wechsler AL, Catovsky D. Splenic angiosarcoma following chemotherapy for follicular lymphoma. Hum Pathol. 1986;17(5):528-30. http://dx.doi.org/10.1016/S0046-8177(86)80044-2. PMid:3516861.
Hara T, Tsurumi H, Kasahara S, et al. Long-term survival of a patient with splenic angiosarcoma after resection, high-dose chemotherapy, and autologous peripheral blood stem cell transplantation. Intern Med. 2010;49(20):2253-7. http://dx.doi.org/10.2169/internalmedicine.49.3969. PMid:20962445.
Koontz BF, Miles EF, Rubio MA, et al. Preoperative radiotherapy and bevacizumab for angiosarcoma of the head and neck: two case studies. Head Neck. 2008;30(2):262-6. http://dx.doi.org/10.1002/hed.20674. PMid:17685450.
Miura H, Asada Y. Treatment of eyelid lesion of angiosarcoma with facial artery recombinant interleukin-2 (rIL-2) injection. J Am Acad Dermatol. 2006;54(5):907-8. http://dx.doi.org/10.1016/j.jaad.2005.11.1047. PMid:16635681.
Takeuchi K, Deguchi M, Hamana S, Motoyama S, Kitazawa S, Maruo T. A case of postirradiation vaginal angiosarcoma treated with recombinant interleukin-2 therapy. Int J Gynecol Cancer. 2005;15(6):1163-5. http://dx.doi.org/10.1111/j.1525-1438.2005.00268.x. PMid:16343203.
Skubitz KM, Haddad PA. Paclitaxel and pegylated-liposomal doxorubicin are both active in angiosarcoma. Cancer. 2005;104(2):361-6. http://dx.doi.org/10.1002/cncr.21140. PMid:15948172.
Amaya CN, Perkins M, Belmont A, et al. Non-selective beta blockers inhibit angiosarcoma cell viability and increase progression free- and overall-survival in patients diagnosed with metastatic angiosarcoma. Oncoscience. 2018;5(3-4):109-19. PMid:29854879.
Notes
Author notes
Correspondence Matheus Dalben Fiorentino Internal Medicine Department - School of Medicine - Universidade de São Paulo (USP) Av. Dr. Enéas de Carvalho Aguiar, 255 – Cerqueira César – São Paulo/SP – Brazil CEP: 05403-000 Phone: +55 (11) 2661-0000 matheus.dalben@outlook.com
Conflict of interest declaration