INTRODUCTION
Neurofibrosarcoma (NFS), also known as malignant schwannoma or peripheral nerve sheath malignancy, is a rare malignant neoplasm of the head and neck region and accounts for 8% to 16% of all cases.1 Its origin is varied. Some may stem from cells of peripheral nerves,2 develop,3 or arise from malignant transformations of preexisting neurofibromas.1,3 It is estimated that 50% of the cases are diagnosed in patients with neurofibromatosis type 1 (NF-1), which is also known as von Recklinghausen's disease.1,3-5 Neurofibromatosis (NF) is an autosomal dominant, high-penetrance disease with a varied expression, with NF-1 being the most common, accounting for 85% to 90% of cases. It is considered that 10% of patients with NF-1 develop NFS.5 The treatment of choice for NFS is surgery, with or without radio- and chemotherapy. The prognosis is usually poor and may present locoregional or distant metastases, mainly to the lungs.6
NFS features are heterogeneous. Therefore, the data provided by the clinical examination are important, especially the presence of neurofibromas or other characteristics of the NF-1, thus directing the diagnostic hypotheses.
CASE REPORT
A 14-year-old girl presented with the chief complaint of “increasing of neck volume and tooth mobility” with intense pain and left lower lip paresthesia over the last 6 months. During the anamnesis, the mother reported that the patient had NF-1, diagnosed in childhood, but did not present the classical signs of neurofibromas, macules, or Lisch nodules.
On examination, facial asymmetry was observed because of an increased volume of the mandible and the left submandibular region, which extended from the chin to the angle of the mandible. The skin was normal, and the regional lymph nodes presented inflammatory features. The intraoral examination of the corresponding lesion revealed an erythematous bulging lesion of the buccal and lingual cortical bone, with central necrosis in the left posterior mandible that extended from the canine to the second molar. Easy bleeding occurs on tooth dislocation (Figure 1).
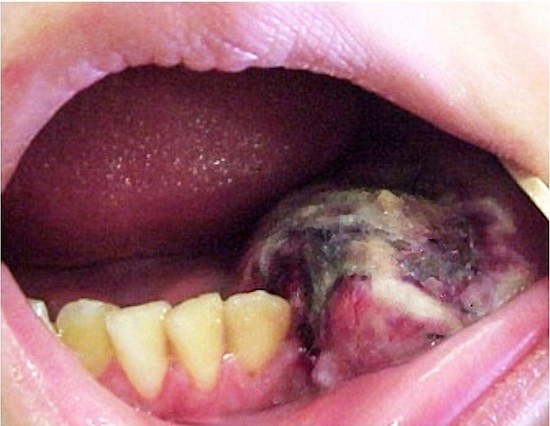 Figure 1
Intraoral examination shows a bulging lesion with central necrosis erupting the buccal and lingual cortical bone.
Figure 1
Intraoral examination shows a bulging lesion with central necrosis erupting the buccal and lingual cortical bone.
Computed tomography revealed the rupture of the lingual and buccal cortical bone. The tumor was a solid lesion with a density similar to that of the soft tissues (Figure 2A and 2B). Similarly, magnetic resonance imaging (MRI) presented the involvement of the buccal and lingual lesion (Figure 2C and 2D).
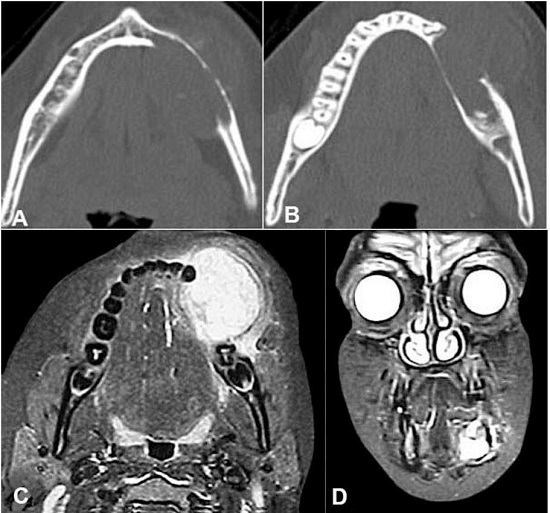 Figure 2
A and B - Axial computed tomography of the mandible revealing the rupture of the lingual and buccal cortical bone. C and D - Facial magnetic resonance imaging C axial plane and D coronal plane. Images weighted in T2 revealing the size and the projection of the buccal and lingual lesion.
Figure 2
A and B - Axial computed tomography of the mandible revealing the rupture of the lingual and buccal cortical bone. C and D - Facial magnetic resonance imaging C axial plane and D coronal plane. Images weighted in T2 revealing the size and the projection of the buccal and lingual lesion.
The three-dimensional reconstruction clearly illustrates the tumor extent, showing irregular bone resorption and destruction of the basal mandible (Figure 3).
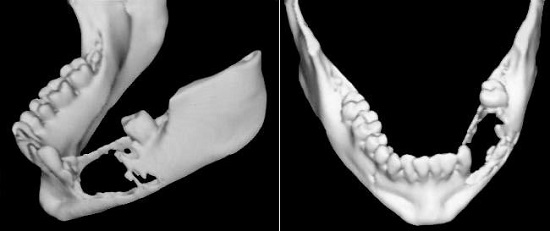 Figure 3
Three-dimensional reconstruction of the mandible illustrating the tumor extent with irregular bone resorption.
Figure 3
Three-dimensional reconstruction of the mandible illustrating the tumor extent with irregular bone resorption.
Due to these clinical features, and the patient’s previous diagnosis of NF-1, the hypothesis of NFS was established and an incisional biopsy was performed. The anatomopathological examination showed a tumor with high cellularity, composed of irregularly organized spindle cells with occasional “spine” and some “Verocay-like” forms, arranged in loose and myxoid areas in an abrupt transition. The nuclei were intensely atypical and exhibited hyperchromatism, pleomorphism, and occasional mitotic figures. There was necrosis in up to 20% of the tumor extent (Figure 4).
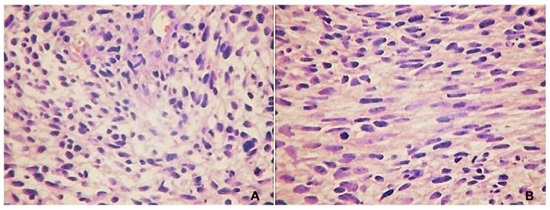 Figure 4
Photomicrographs of the tumor. A - Cellular arrangements in loose bundles, perivascular disposition, and areas of myxoid aspect. The cells are elongated, with poorly defined borders, and the nuclei are equally elongated and atypical (HE, 400×). B - Cellular arrangements in bundles with occasional “fishbone” arrangement, fibrosarcoma-like. Note the presence of mitoses (HE, 400×).
Figure 4
Photomicrographs of the tumor. A - Cellular arrangements in loose bundles, perivascular disposition, and areas of myxoid aspect. The cells are elongated, with poorly defined borders, and the nuclei are equally elongated and atypical (HE, 400×). B - Cellular arrangements in bundles with occasional “fishbone” arrangement, fibrosarcoma-like. Note the presence of mitoses (HE, 400×).
The immunohistochemical panel was characterized by the focal positivity for vimentin, HHF-35, and p53 actin and negativity for S-100, desmin, and CD99 (Figure 5). The Ki-67 was positive in 95% to 98% of the tumor cells.
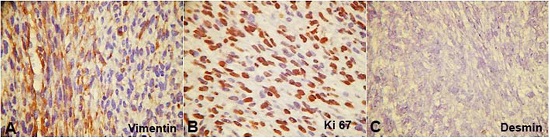 Figure 5
Photomicrographs of the tumor. A - Streptavidin-biotin-peroxidase. Mayer’s hematoxylin and diaminobenzidine. Occasional vimentin positivity. B - Streptavidin-biotin-peroxidase. Mayer’s hematoxylin and diaminobenzidine. Ki-67 proliferative index. C - Streptavidin-biotin-peroxidase. Mayer’s hematoxylin and diaminobenzidine. Negativity to desmin.
Figure 5
Photomicrographs of the tumor. A - Streptavidin-biotin-peroxidase. Mayer’s hematoxylin and diaminobenzidine. Occasional vimentin positivity. B - Streptavidin-biotin-peroxidase. Mayer’s hematoxylin and diaminobenzidine. Ki-67 proliferative index. C - Streptavidin-biotin-peroxidase. Mayer’s hematoxylin and diaminobenzidine. Negativity to desmin.
The final diagnosis of NFS was established by associating the clinical, imaging, and pathological data. The treatment of choice was neoadjuvant chemotherapy and segmental mandibulectomy (Figure 6). After surgery, chemotherapy and adjuvant radiotherapy were performed. The patient has been in the follow-up for 10 years, with no evidence of recurrence.
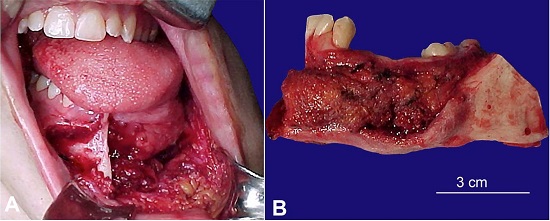 Figure 6
A - Intraoperative view of the segmental mandibulectomy. B - Resected surgical.
Figure 6
A - Intraoperative view of the segmental mandibulectomy. B - Resected surgical.
DISCUSSION
NFS may present unusual clinical and imaging features.1 The clinical characteristic of NFS is its rapid growth, either asymptomatic or painful, accompanied by paresthesia, muscular atrophy, and weakness. The tumor margins are irregular, and there may be ulcerations on the surface.3 In this case, the patient complained of pain and lip paresthesia, and the surface was ulcerated.
The mandible, lip, and buccal mucosa are the most commonly involved sites in the oral cavity affected by NFS.1 Intraosseous NFS of the mandible may present with an increased volume of the mandibular canal or mental foramen.7,8 In some cases, the radiographic findings suggest malignancy as an irregularity of the margins of the lesion.7 In our case, we could observe the extensive destruction of the mandibular base, the loss of the mandibular canal definition, and the ill-defined margins observed on imaging examinations.
Histologically, NFS is composed of spindle cells, with elongated, rounded, or bulky nuclei, presenting hyperchromatism and varying degrees of pleomorphism.1,3 The stroma is usually fibrotic or myxomatous where heterotrophic elements such as mature bone or cartilaginous islets, skeletal muscle, glands and sites of hemorrhage, and necrosis can be found.3 The presence of spindle cells with atypical nuclei in our case was consistent with a lesion of malignant mesenchymal origin, that is, a sarcoma. The final diagnosis of NFS by conventional methods of anatomic-pathological examination is difficult to establish1 because the characteristics are similar to those of other sarcomas that present spindle cell proliferation such as fibrosarcoma, leiomyosarcoma, monophasic synovial sarcoma, epithelioid sarcoma, and malignant fibrous histiocytoma.1,5,7 Usually, the only histological distinction between these sarcomas and NFS is their origin of nerve branches,9 which was not observed in our case.
The precise diagnosis can be established with the aid of the electron microscopy and immunohistochemistry. The biological marker mostly used is S-100, which is positive in 17% to 56% of cases of NFS.10,11 It is a specific protein for nerve tissue12 and is observed in numerous cells originating from the neural crest, including Schwann cells.1 The immunohistochemical panel of this case showed negativity for the S-100 protein, which as already mentioned, is negative in approximately 50% of cases; focal positivity for the intermediate vimentin filament, present in fibroblasts, demonstrating the mesenchymal origin of the tumor; and negativity for desmin present in smooth and skeletal muscles, thus ruling out the muscular origin of the tumor. Positivity for the Ki-67 protein and proliferative index of 95% to 98% demonstrated rapid growth and aggressiveness of the tumor. The final diagnosis was established as NFS according to the morphologic features and immunohistochemical panel.
There are different points of view regarding the treatment of NFS. Radical surgery is imperative1,3, and complementary radiotherapy increases the patient's survival rate by 5 years.13,14 Radiotherapy may also be used in cases of local recurrence and inoperable tumors.3 Some authors argue that using radiotherapy as the first treatment method leads to poor results15 and supports postsurgical radiation.16 In a study17 of head and neck sarcomas, the authors proposed combined surgery and radiotherapy in cases of advanced tumors (larger than 5 cm), whereas they opted for only surgery for lesions of smaller sizes. In cases of total irresectability of the tumor, palliative radiotherapy and chemotherapy sessions were provided.17 In some studies13,18, the indication of chemotherapy is controversial. The patient was initially treated with neoadjuvant chemotherapy to reduce the lesion size, followed by radical surgery with free margins, associated with radiotherapy and chemotherapy. This method was chosen based on the tumor extent (larger than 5 cm), its aggressive behavior with high Ki-67, and its high indices of locoregional and distant recurrences and metastases, which represents approximately 33% of cases.16,19
High rates of recurrence following multimodality therapy in early disease, low response rates to cytotoxic chemotherapy in advanced disease, and propensity for rapid disease progression and high mortality19 make the prognosis of NFS poor19-21, with a 5-year survival rate ranging from 15% to 30%21 for patients with NF-1. Locoregional or distant metastasis may occur4,5 mainly in the lungs.6 The lymph nodes are rarely affected.1,6,14 The development of new, worsening, or persistent pain in the neurofibroma of a patient with NF-1 is an important symptom that should always be conscientiously evaluated.19 Our patient has been followed up for 10 years, with no evidence of recurrence or metastasis, possibly because of better investigation and fast intervention at earlier stages of disease in patients with NF-1 due to improvements in imaging and diagnostic techniques.
NFS may present different clinical and imaging features, with MRI being the most useful imaging modality for characterizing the anatomical extent of the tumor.19 The biopsy is fundamental for the final diagnosis. Despite the fact that anatomopathological presentation is very similar to other sarcomas, immunohistochemistry is usually necessary to confirm the diagnosis. In the present case, knowing that the patient had NF-1, NFS was the initial working diagnosis.
The patient’s mother signed the consent declaration after this research was approved by the Research Ethical Committee of the Hospital.
 Juliane Pirágine Araujo
Juliane Pirágine Araujo
