Article / Autopsy Case Report
A second reported malignancy in a patient with Morquio syndrome
A second reported malignancy in a patient with Morquio syndrome
Autopsy and Case Reports, vol. 7, no. 2, pp. 9-14, 2017
Hospital Universitário da Universidade de São Paulo
Received: 05 April 2017
Accepted: 02 June 2017
ABSTRACT: Morquio syndrome is a rare lysosomal storage disease that affects multiple organ systems. However, it is rarely associated with malignancy. We present the case of a 30-year old man with Morquio syndrome associated with gastric adenocarcinoma. This case also demonstrates two other findings that have not been previously described in patients with Morquio syndrome - malrotation of brainstem and cerebellum, without clinical neurologic deficit, and persistence of fetal lobulation in the kidneys.
Keywords: Lysosomal storage diseases, Mucopolysaccharidosis IV, Stomach Neoplasm.
INTRODUCTION
Morquio syndrome is a rare autosomal recessive lysosomal storage disease referred to as mucopolysaccharidosis (MPS) type IV, which is further divided into IVA and IVB subtypes depending on the enzyme deficiency. Type IVA is caused by the deficiency of enzyme N-acetyl-galactosamine-6-sulfatase causing intracellular accumulation of keratan sulfate and chondroitin 6-sulfate whereas type IVB is caused by β-galactosidase deficiency causing accumulation of keratan sulfate only.1 Morquio patients can present with musculoskeletal abnormalities such as muscular atrophy, pectus carinatum, dysostosis multiplex, odontoid hypoplasia, short stature, dorsilumbar kyphoscoliosis and waddling gait. Atlantoaxial subluxation leading to spinal cord compression in the upper cervical region can be life threatening. This subluxation is a consequence of odontoid dysplasia, which is a major complication of MPS type IV.2 The manifestations that can develop later in the life include corneal opacity, deafness, hypermobility of joints, cardiac abnormalities, quadriplegia and respiratory paralysis.3 Morquio syndrome has rarely been associated with carcinomas or other malignancies. A search of the literature revealed only one case of osteosarcoma arising in the vicinity of hardware in the femur of an 18-year-old man with Morquio syndrome.4 We present a case of metastatic gastric adenocarcinoma arising in a man with Morquio syndrome, which was diagnosed in early childhood and presented the typical findings of this syndrome at the end of life.
CASE REPORT
The patient was a 30-year-old African American man with Morquio syndrome diagnosed at age 3 by a constellation of physical findings and laboratory investigation. He had classic physical findings of Morquio syndrome including coarse facial features, short stature, short neck, pectus carinatum, genu valgum and thoracic kyphosis. Radiographic studies also revealed findings consistent with Morquio syndrome. However, records of pertinent laboratory testing, such as increased urinary glycosaminoglycan levels or GALNS gene sequencing, were not available for review. The family history was significant for breast carcinoma in father, skin carcinoma in sister and esophageal carcinoma in paternal grandfather. He was diagnosed with poorly differentiated gastric adenocarcinoma at age 28 and was being treated with chemo and radiotherapy. The disease however persisted, and later the patient returned to the hospital complaining of back pain and shortness of breath. At the time of admission, the man was dyspneic with normal vital signs. Physical examination revealed a body mass index of 19, cachectic appearance, anasarca, pectus carinatum, genu valgum, short stature and thoracic kyphosis with exaggerated lumbar lordosis (Figure 1A, 1B and 1C). However, no neurological deficit was noted.
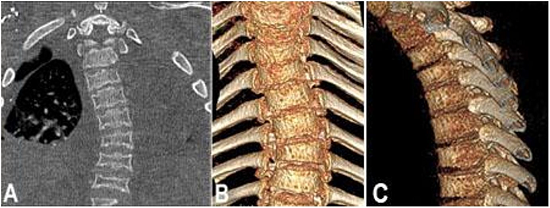
Figure 1
Thoracic CT Scan showing scoliosis of the thoracic spine (A, coronal plane) and 3D reconstruction showing thoracic scoliosis and kyphosis (B and C, coronal and sagittal planes).
Laboratory studies were within normal limits except for anemia with hemoglobin of 8.5 gm/dL (normal range [NR]; 13.5 to 18.0 gm/dL) and leukopenia with a white blood cell count of 1700/mm3 [NR]; 4000-11000/mm3). Imaging studies revealed cardiomegaly, multifocal osseous metastatic disease (Figure 2), degenerative disc disease resulting in varying degrees of central canal and foraminal stenosis, wedge deformities of C2 and C4 without evidence of displaced fracture, chronic osseous changes of upper lumbar spine with significant vertebral body compression, bilateral hip disarticulations and small liver metastases.
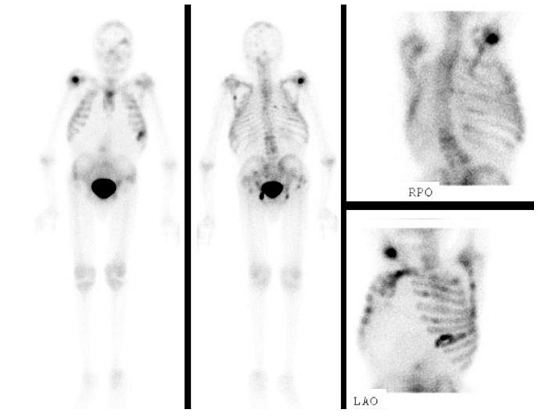
Figure 2
Bone scintigraphy using 99mTc in anterior, posterior, right posterior oblique, and left anterior oblique views showing multifocal osseous metastatic disease.
His condition deteriorated during the course of hospitalization and on hospital day 10 he was found unresponsive in pulseless electrical activity and was unable to be resuscitated following 24 minutes of attempted resuscitation. He was pronounced dead and a postmortem examination was requested.
AUTOPSY FINDINGS
Significant findings at autopsy included poorly differentiated gastric adenocarcinoma (Figure 3A) metastatic to left supraorbital dura, pituitary gland, pericardium, lungs with evidence of lymphangitic carcinomatosis, liver, peripancreatic soft tissue, mesentery of large bowel, wall of colon, bilateral periadrenal soft tissue, thoracic vertebrae, periprostatic soft tissue and urinary bladder. Additionally, the pericardium, peritoneum, peripancreatic soft tissue, periadrenal soft tissue, perithyroidal soft tissue, laryngeal soft tissue, periprostatic soft tissue, small and large bowel and subdural soft tissue surrounding the spinal cord showed mucoid material containing distended cells with foamy cytoplasm and eccentric nuclei (Figure 3B and 3C), consistent with a mucopolysaccharidosis. Other notable findings at autopsy were persistence of fetal lobulation in each kidney, severe malrotation of brainstem and cerebellum (Figure 4A and 4B) and thinning of optic chiasma and left optic nerve.
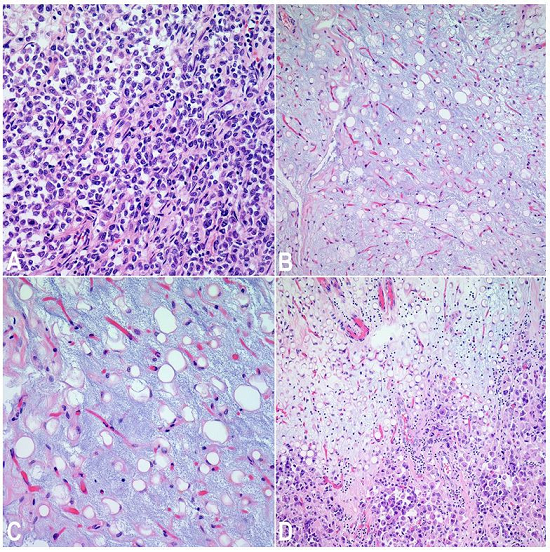
Figure 3
Photomicrography of poorly differentiated gastric adenocarcinoma (A, H&E, 400X) and mucoid material consistent with mucopolysaccharide material (B and C, H&E, 200X and 400X respectively). Metastatic tumor in a background of mucoid material (D, H&E, 200x).
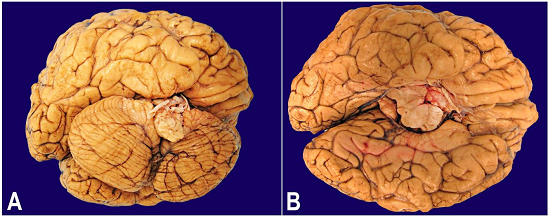
Figure 4
Macroscopic examination of brain revealed malrotation of cerebellum (A) and brainstem (B).
Death was likely due to aggressive metastatic gastric adenocarcinoma, with skeletal complications of probable MSP IV - namely vertebral scoliosis and kyphosis as well as atlantoaxial instability - likely contributing to death, as well. The manner of death was natural.
DISCUSSION
Lysosomes are the key component of intracellular digestion. They contain hydrolytic enzymes which are secreted into intracellular organelles and catalyze the breakdown of complex macromolecules. With inherited deficiency of lysosomal enzymes, the catabolism of the macromolecules remains incomplete leading to accumulation of insoluble metabolite in the lysosomes. The lysosomes therefore become larger and interfere with normal cell function leading to “lysosomal storage diseases”.5 It is not just the enzyme deficiency but several other defects that lead to lysosomal storage diseases, including deficiency of enzyme activator, deficiency of substrate activating protein, and deficiency of transport protein facilitating exit of digested material from lysosome.6
MPS represent a subset of lysosomal storage diseases in which mucopolysaccharides / glycosaminoglycans (GAGs) accumulate in the lysosomes. GAGs include chondroitin-4-sulfate, chondroitin-6-sulfate, heparan sulfate, dermatan sulfate, keratan sulfate and hyaluronan. MPS are usually progressive diseases and involve multiple organ systems. Each type of MPS has a unique clinical presentation, however, most are associated with coarse facial features, corneal clouding, joint stiffness, skeletal deformities, hepatosplenomegaly, valvular lesions and lesions of the brain.
Morquio syndrome (MPS IV) has an incidence of 1 in 75,000.7 It is further divided into IVA and IVB subtypes depending on the enzyme deficiency. Type IVA is caused by deficiency of enzyme N-acetyl-galactosamine-6-sulfatase and type IVB is caused by β-galactosidase deficiency.1 Clinical presentation and other features of Morquio syndrome are summarized in Table 1

Lysosomes play a vital role in biology of malignant cells. Their effect can be oncogenic or on the contrary may also be apoptotic.8 This occurs via complex molecular pathways involving cathepsins and other proteins. Regarding MPS associated with neoplasia, Gaucher disease is the disorder that has been studied most extensively. Carcinomas of breast, prostate, colon, lung, ovary, kidney, bladder, liver and thyroid, non-melanoma skin cancers, ganglioneuroma and neurosarcoma, angiosarcoma, testicular rhabdomyosarcoma and carcinoid tumor have all been reported in patients with Gaucher disease.9 Hematologic malignancies including multiple myeloma, Hodgkin and non-Hodgkin lymphoma, acute lymphoblastic and myeloid leukemias, and chronic lymphocytic leukemia have also been reported in patients with Gaucher disease.9 Gaucher disease, for example, confers a relative risk of 5.9 of developing multiple myeloma.9 Overall, though, with the exception of Gaucher disease, lysosomal storage diseases in general and MPS in particular are rarely associated with malignancy. This may be secondary to the decreased life expectancy of patients with many MPS, or to other factors which have yet to be understood through further investigation and research. Thorough search of literature revealed only rare cases of other MPS associated with different malignancies, which are summarized in Table 2 and include the present case.

Our case showed aggressive metastatic gastric adenocarcinoma in a patient with likely MPS IV based on clinical physical and laboratory findings in the past and characteristic findings at postmortem examination. In 9 out of 11 metastatic sites, the metastatic adenocarcinoma was present in a background of mucoid material containing distended cells with foamy cytoplasm and eccentric nuclei consistent with likely underlying mucopolysaccharidosis (Figure 3D). This finding suggests a causal relationship at least between metastatic propensity of a tumor and MPS such as MPS IV. However the molecular mechanisms underlying such a potential relationship need to be elucidated to confirm such a relationship, if one exists at all.
Another notable finding in the current case was persistent fetal lobulation in the kidneys, which represent a normal variant reflecting incomplete fusion of the developing renal lobules. The kidney function is essentially normal, however, the ultra-sonographic findings in such kidneys may be mistaken as scars.13 To our limited knowledge, this finding has not been described in patients with Morquio syndrome. Lastly, the brain examination of our case also demonstrated a severe malrotation of brainstem and cerebellum. However, the patient did not have any neurologic deficit in 30 years of life. It is highly likely that this malrotation was related to odontoid hypoplasia and could very well be a protective mechanism against brainstem and spinal cord compression, which is common complication of odontoid hypoplasia in Morquio patients.
CONCLUSION
Morquio syndrome is a well-known entity but our understanding of the disorder is still limited. This case elaborates a few new findings in patients with MPS IV. Take home messages from the case are: i) patients with Morquio syndrome may have persistence of fetal lobulations in kidneys, which should not necessarily be interpreted as renal scarring on ultrasound examinations; ii) neurologic abnormalities in MPS include structural brain abnormalities such as malrotation of brainstem and cerebellum and such findings can be asymptomatic; and iii) MPS may play a role in carcinogenesis and/or metastasis of epithelial neoplasms.
REFERENCES
1 Matalon R, Arbogast B, Dorfman A. Deficiency of chondroitin sulfate N-acetylgalactosamine 4-sulfate sulfatase in Maroteaux-Lamy syndrome. Biochem Biophys Res Commun. 1974;61(4):1450-7. 4218107 http://dx.doi.org/10.1016/S0006-291X(74)80446-8.
2 Nelson J, Kinirons M. Clinical findings in 12 patients with MPS IV A (Morquio’s disease). Further evidence for heterogeneity. Part II: dental findings. Clin Genet. 1988;33(2):121-5. 3129222 http://dx.doi.org/10.1111/j.1399-0004.1988.tb03422.x.
3 Jenkins P, Davies GR, Harper PS. Morquio Brailsford disease: a report of four affected sisters with absence of excessive keratan sulphate in the urine. Br J Radiol. 1973;46(549):668-75. 4199240 http://dx.doi.org/10.1259/0007-1285-46-549-668
4 Massachusetts Medical Society. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 4-1991 — An 18-Year-Old Man with Morquio’s Syndrome and a Tumor of the Right Femur. N Engl J Med. 1991;324(4):251-9. http://dx.doi.org/10.1056/NEJM199101243240408. PMid:1898672
5 Wraith JE. Lysosomal disorders. Semin Neonatol. 2002;7(1):75-83. 12069540 http://dx.doi.org/10.1053/siny.2001.0088
6 Tager JM. Inborn errors of cellular organelles: an overview. J Inherit Metab Dis. 1987;10(Suppl 1):3-10. 3119939 http://dx.doi.org/10.1007/BF01812842
7 Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet. 1997;101(3):355-8. 9439667 http://dx.doi.org/10.1007/s004390050641
8 Appleqvist H, Wäster P, Kågedal K, Öllinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol. 2013;5(4):214-26. 23918283 http://dx.doi.org/10.1093/jmcb/mjt022
9 Rosenbloom BE, Weinreb NJ, Zimran A, Kacena KA, Charrow J, Ward E. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood. 2005;105(12):4569-72. 15718419 http://dx.doi.org/10.1182/blood-2004-12-4672
10 Lo SM, Stein P, Mullaly S, et al. Expanding spectrum of the association between type 1 Gaucher disease and cancers: a series of patients with up to 3 sequential cancers of multiple types - correlation with genotype and phenotype. Am J Hematol. 2010;85(5):340-5. http://dx.doi.org/10.1002/ajh.21684. PMid:20425796
11 McGovern MM, Lippa N, Bagiella E, Schuchman EH, Desnick RJ, Wasserstein MP. Morbidity and mortality in type B Niemann-Pick disease. Genet Med. 2013;15(8):618-23. 23412609 http://dx.doi.org/10.1038/gim.2013.4
12 Stockler S, Kleinert R, Ebner F, Paschke E. Mucopolysaccharidosis I and intracranial tumor in a patient with high-pressure hydrocephalus. Pediatr Radiol. 1993;23(5):353-4. 8233684 http://dx.doi.org/10.1007/BF02011955
13 Patriquin H, Lefaivre JF, Lafortune M, Russo P, Boisvert J. Fetal lobation. An anatomo-ultrasonographic correlation. J Ultrasound Med. 1990;9(4):191-7. 2184242 http://dx.doi.org/10.7863/jum.1990.9.4.191
Notes
Author notes
Correspondence Ameer Hamza Department of Pathology - St John Hospital and Medical Center 22101 Moross Road - Detroit/MI - United States of America 48236 Phone: +1 (313) 613-7511 / Fax: +1 (313) 343-8318 ameerhamza7@hotmail.com
Conflict of interest declaration