Article / Autopsy Case Report
A rare case of centronuclear myopathy with DNM2 mutation: genotype-phenotype correlation
A rare case of centronuclear myopathy with DNM2 mutation: genotype-phenotype correlation
Autopsy and Case Reports, vol. 7, no. 2, pp. 43-48, 2017
Hospital Universitário da Universidade de São Paulo
Received: 26 April 2017
Accepted: 29 May 2017
ABSTRACT: Centronuclear myopathy (CNM) is a group of rare genetic muscle disorders characterized by muscle fibers with centrally located nuclei. The most common forms of CNM have been attributed to X-linked recessive mutations in the MTM1 gene; autosomal-dominant mutations in the DNM2 gene-encoding dynamin-2, the BIN1 gene; and autosomal-recessive mutations in BIN1, RYR1, and TTN genes. Dominant CNM due to DNM2 mutations usually follows a mild clinical course with the onset in adolescence. Currently, around 35 mutations of the DNM2 gene have been identified in CNM; however, the underlying molecular mechanism of DNM2 mutation in the pathology of CNM remains elusive, and the standard clinical characteristics have not yet been defined. Here, we describe the case of a 17-year-old female who presented with proximal muscle weakness along with congenital anomalous pulmonary venous connection (which has not been described in previous cases of CNM), scoliosis, and lung disease without a significant family history. Her creatine kinase level was normal. Histology, special stains, and electron microscope findings on her skeletal muscle biopsy showed CNM with the characteristic features of a DNM2 mutation, which was later confirmed by next-generation sequencing. This case expands the known clinical and pathological findings of CNM with DNM2 gene mutation.
Keywords: Centronuclear, Myopathy, DNM2, Congenital.
INTRODUCTION
Centronuclear myopathy (CNM) is a rare congenital myopathy, which was first described as myotubular myopathy by Spiro et al.1 in 1966. The muscle biopsy is characterized by myofibers with centrally placed nucleoli, which are reminiscent of the myotube stage of muscle development. The authors suggested that it originated from an arrested maturation of embryonic muscle. Hence, the condition was called myotubular myopathy. It remains unclear if muscle development was arrested; therefore the term “centronuclear myopathies” was preferred, especially for the late onset forms. The term “myotubular” is now used to denote severe infantile cases, and reserve CNM denotes clinically milder cases affecting older patients in whom the muscle biopsy tends to appear “more mature.”2-4
CNM is usually caused by a mutation in the myotubularin (MTM1), amphiphysin 2 (BIN1), and dynamin 2 (DNM2) genes, which are involved in membrane remodeling and trafficking, suggesting a common CNM pathophysiology.5 In addition to CNM, dissimilar DNM2 mutations are associated with Charcot-Marie-Tooth (CMT) peripheral neuropathy (CMTD1B and CMT2M), indicating a tissue-specific impact of the mutations. Defects in membrane trafficking due to DNM2 mutations potentially represent a common pathological mechanism in CNM and CMT.6
Three main forms of CNM are recognized according to the mode of inheritance and clinical presentation7 (Table 1):
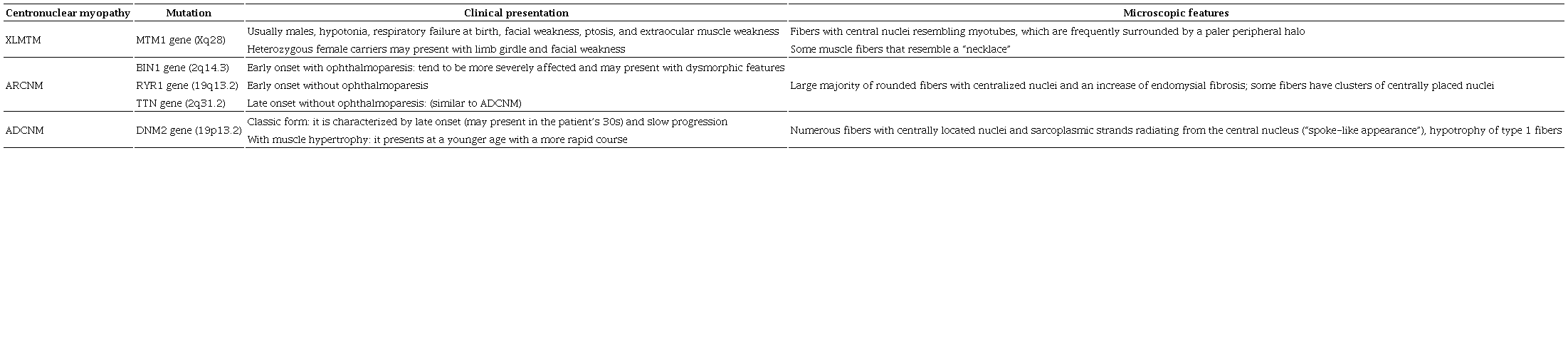
ADCNM = autosomal dominant centronuclear myopathy; ARCNM = autosomal recessive centronuclear myopathy; XLMTM = X-linked myotubular myopathy.
- 1.
1
X-linked myotubular myopathy (XLMTM): This form of CNM is the most severe and the most commonly diagnosed type, which is caused by a mutation in the MTM1 gene located at Xq27-q28.4,8 Almost all cases occur in males and present with severe hypotonia, which frequently causes respiratory failure at birth. Facial weakness, ptosis, and extraocular muscle weakness are common. Rarely, abnormal genital development occurs.9 Heterozygous female carriers may present with limb girdle and facial weakness.10 Mutations in MTM1 have also been recognized as the underlying cause of atypical forms of XLMTM in newborn boys, female infants, and adult men and women, which presents a histological alteration in some muscle fibers that resemble a necklace (“necklace fibers”).11 Muscle biopsies from XLMTM patients show numerous fibers with central nuclei resembling myotubes, which are frequently surrounded by a paler peripheral halo. These perinuclear paler halos are due to mitochondria aggregates, glycogen granules, and a reduction in myofilaments.12,13
- 2.
2
Autosomal recessive CNM (ARCNM): This form presents with relatively mild weakness that may be unrecognized in the neonatal period. It is caused by mutations in the bridging integrator 1 gene (BIN1).14,15 Mutations in the RYR1 gene have also been described in patients with congenital myopathy with central nuclei, although it is now recognized that these patients have a form of RYR1-related congenital myopathy rather than CNM.16,17 Recessive mutations in TTN that encode the giant sarcomeric ruler protein, titin, have also been identified in some patients with congenital myopathies with central nuclei.18 The muscle biopsies in BIN1-related CNM show a large majority of rounded fibers with centralized nuclei and an increased amount of endomysial fibrosis; some fibers have clusters of centrally placed nuclei on H&E staining. The presence or absence of ophthalmoparesis in early onset cases has been suggested as a means to clinically classify ARCNM into three subgroups:19 (i) early onset with ophthalmoparesis: patients tend to be more severely affected and may present with dysmorphic features; (ii) early onset without ophthalmoparesis; and (iii) late onset without ophthalmoparesis: affected patients have a clinical phenotype similar to patients with autosomal dominant CNM.
- 3.
3
Autosomal dominant CNM (ADCNM): This disorder not only involves mainly limb girdle, trunk, and neck muscles, but also it may affect the distal muscles. Weakness may not become evident until the third decade of life. It is caused by a mutation in dynamin 2 (DNM2) on chromosome 19.20 The most prominent histopathological features include the frequency of centrally located nuclei in a large number of the extrafusal muscle fibers, the radial arrangement of sarcoplasmic strands around the central nuclei, and the predominance and hypotrophy of type 1 fibers. ADCNM can be divided to two subgroups due to the presence or absence of muscle hypertrophy:19 (i) classic form: this is characterized by late onset and slow progression; and (ii) with muscle hypertrophy: this usually presents at a younger age and has a more rapid course.
CASE REPORT
A 17-year-old Caucasian female with a past medical history of anomalous pulmonary venous connection (left to right shunt), biphasic thoracolumbar scoliosis, and restrictive lung disease, presented with progressive proximal muscle weakness and lumbar pain. There was no specific family history of neurological disease. Upon physical examination, we observed levoscoliosis and weakness in the bilateral upper extremities (4/5) and the lower extremities (3/5). She was unable to walk on her toes or heels, and was unable to arise from a squatting position, although her gait was not grossly abnormal on observation. There was no facial weakness, and her extraocular movements were intact. Sensory examination showed intact sensation bilaterally. There was no spasticity, Babinski sign, or ankle clonus.
An x-ray showed scoliosis and kyphoscoliosis. Magnetic resonance imaging findings were suggestive of paraspinal muscle edema suspicious for an underlying myopathy process. A partial anomalous pulmonary venous return with left to right shunt was also observed. An electromyography (EMG) showed electrodiagnostic evidence of diffuse myopathic process. The creatine kinase (CK) level was normal: 68 (reference range: 0-250 U/L).
The microscopic examination of the muscle biopsy (left vastus lateralis) showed excess variability in fiber size (Figure 1A), and frequent myonuclei in the center of the muscle fibers (Figure 1B), which often formed chains when viewed longitudinally (Figure 1C). Degenerating fibers, angulated, split fibers, and nuclear bags were not present. Myophagocytosis was not present, and inflammatory infiltrates were not seen. Rare fibers contained central red stippling; however, definitive ragged red fibers were not seen. NADH histochemistry showed increased reaction in the center of some fibers with radial arrangement of the intermyofibrillar network radiating from the central nucleus (a spoke-like appearance on the fibers) (Figure 1D).
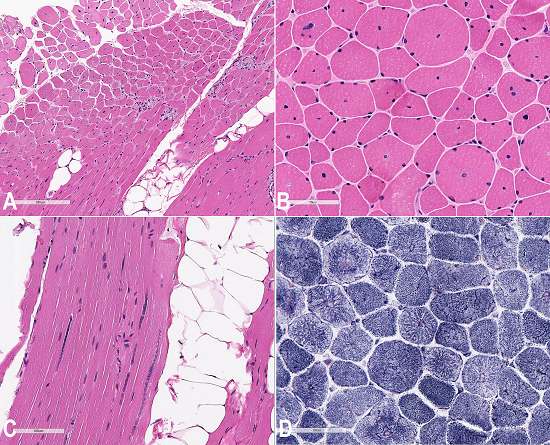
Figure 1
Photomicrography of the muscle biopsy. A - Fiber size variation and centralized nuclei especially in small fibers (H&E); B - Central nuclei are observed in majority of cells (H&E); C - Centrally located nuclei often forming chains in a longitudinal section (H&E); D - Radial arrangement of inter-myofibrillar network (spoke-like appearance) (NADH stain).
ATPase (pH 9.8, 4.6, 4.3) histochemical stains show type I fiber predominating (more than 90%), and type II fibers were normal in size or were hypertrophic (Figure 2A). Electron microscopy with toluidine blue staining showed disruption of myofibers particularly around the central nuclei (Figure 2B). Glycogen content appeared mildly increased in areas, and scattered collections of mitochondria and occasional nonspecific inclusions were noted.
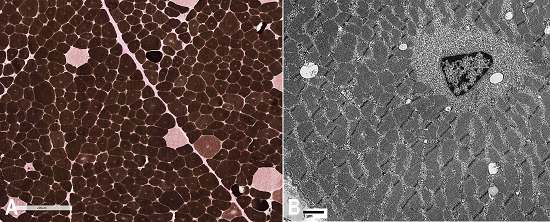
Figure 2
Photomicrography of the muscle biopsy. A - ATPase (pH 4.3) type II fiber hypertrophy and type I fiber predominance (>90%); B - Electronic microscopy: fibers with central nuclei and radial sarcoplasmic strands (X 6300).
Mitochondrial DNA (mtDNA) whole genome sequencing was performed at a third-party lab (using the internal guidelines at the Medical Neurogenetic Lab, State of Georgia license #044-146) and showed a heterozygous missense variant in exon 11 of the DNM2 gene located on chromosome 19; DNM2 (NM_001005360.2):c.1393C>T: p.Arg465Trp. This variant corresponded to the SNP rs121909091 and had been observed previously in at least three individuals with CNM, according to the ClinVar database. It is not found in more than 60,000 controls in the ExAC database.21
DISCUSSION
CNM is a rare congenital myopathy, which can be diagnosed based on histology. Three forms of the disease are clinically recognized: (i) the X-linked severe neonatal form, which is caused by MTM1 mutation; (ii) the autosomal recessive childhood onset form, which is usually caused by BIN1 mutation; and (iii) the autosomal dominant adult-onset form, which is caused by DNM2 mutation. Genotype-phenotype correlation hypotheses are drawn from published and new data and allow an efficient screening strategy for molecular diagnosis. Here, we report the clinical, pathological, and molecular characteristics of a 17-year-old patient with DNM2-related CNM, which was confirmed by genetic testing. The patient presented with muscle weakness and associated disabilities (thoracolumbar scoliosis with restrictive lung disease), as well as anomalous pulmonary venous connection. Because the cardiac abnormality has not been described in patients with DNM2-related CNM, it is not clear whether it could be just a co-occurrence condition or the first description of this new finding. Therefore, more evidence is needed to conclude this matter. The laboratory work-up showed normal blood CK levels; however, the EMG study was consistent with myopathy. The microscopic and histochemistry studies of the muscle biopsy showed radial oxidative staining of sarcoplasmic strands (on NADH staining) and type I muscle fiber predominance with type II muscle fiber hypertrophy (on ATPase staining), which was suggestive of DNM2-related changes. A further genetic work-up revealed a DNM2 mutation on chromosome 19, and confirmed the diagnosis of ADCNM.
REFERENCES
1 Spiro AJ, Shy GM, Gonatas NK. Myotubular myopathy: persistence of fetal muscle in an adolescent boy. Arch Neurol. 1966;14(1):1-14. 4954227 http://dx.doi.org/10.1001/archneur.1966.00470070005001
2 Fardeau M. Skeletal muscle pathology. In: Mastaglia FL, Walton Sir JN, editors. Congenital myopathies. London: Churchill Livingston; 1982. p. 161-203.
3 Fardeau M, Tome F. Congenital myopathies. In: Engel AG, Franzini-Armstrong C, editors. Myology. 2nd ed. New York: McGraw Hill; 1994. p. 1500-25.
4 North KN. Congenital myopathies. In: Engel A, Franzini-Armstrong C, editors. Myology. 3rd ed. New York: McGraw Hill; 2004. p. 1473-533.
5 Toussaint A, Cowling BS, Hnia K, et al. Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. 2011;121(2):253-66. 20927630 http://dx.doi.org/10.1007/s00401-010-0754-2
6 Bohm J, Biancalana V, DeChene ET, et al. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum Mutat. 2012;33(6):949-59. 22396310 http://dx.doi.org/10.1002/humu.22067
7 Zanoteli E, Oliveira AS, Schmidt B, Gabbai AA. Centronuclear myopathy: clinical aspects of ten Brazilian patients with childhood onset. J Neurol Sci. 1998;158(1):76-82. 9667782 http://dx.doi.org/10.1016/S0022-510X(98)00091-4.
8 Laporte J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13(2):175-82. 8640223 http://dx.doi.org/10.1038/ng0696-175
9 Bartsch O, Kress W, Wagner A, Seemanova E. The novel contiguous gene syndrome of myotubular myopathy (MTM1), male hypogenitalism and deletion in Xq28: report of the first familial case. Cytogenet Cell Genet. 1999;85(3-4):310-4. 10449925 http://dx.doi.org/10.1159/000015284
10 Sutton IJ, Winer JB, Norman AN, Liechti-Gallati S, MacDonald F. Limb girdle and facial weakness in female carriers of X-linked myotubular myopathy mutations. Neurology. 2001;57(5):900-2. 11552027 http://dx.doi.org/10.1212/WNL.57.5.900
11 Bevilacqua JA, Bitoun M, Biancalana V, et al. “Necklace” fibers, a new histological marker of late-onset MTM1-related centronuclear myopathy. Acta Neuropathol. 2009;117(3):283-91. 19084976 http://dx.doi.org/10.1007/s00401-008-0472-1
12 Romero NB. Centronuclear myopathies: a widening concept. Neuromuscul Disord. 2010;20(4):223-8. 20181480 http://dx.doi.org/10.1016/j.nmd.2010.01.014
13 Pierson CR, Tomczak K, Agrawal P, Moghadaszadeh B, Beggs AH. X-linked myotubular and centronuclear myopathies. J Neuropathol Exp Neurol. 2005;64(7):555-64. 16042307 http://dx.doi.org/10.1097/01.jnen.0000171653.17213.2e
14 Nicot AS, Toussaint A, Tosch V, et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39(9):1134-9. 17676042 http://dx.doi.org/10.1038/ng2086
15 Claeys KG, Maisonobe T, Böhm J, et al. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology. 2010;74(6):519-21. 20142620 http://dx.doi.org/10.1212/WNL.0b013e3181cef7f9
16 Jungbluth H, Zhou H, Sewry CA, et al. Centronuclear myopathy due to a de novo dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. 2007;17(4):338-45. 17376685 http://dx.doi.org/10.1016/j.nmd.2007.01.016
17 Bevilacqua JA, Monnier N, Bitoun M, et al. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalisation and large areas of myofibrillar disorganisation. Neuropathol Appl Neurobiol. 2011;37(3):271-84. 21062345 http://dx.doi.org/10.1111/j.1365-2990.2010.01149.x.
18 Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. 2013;81(14):1205-14. 23975875 http://dx.doi.org/10.1212/WNL.0b013e3182a6ca62
19 Jeannet PY, Bassez G, Eymard B, et al. Clinical and histologic findings in autosomal centronuclear myopathy. Neurology. 2004;62(9):1484-90. 15136669 http://dx.doi.org/10.1212/01.WNL.0000124388.67003.56
20 Bitoun M, Maugenre S, Jeannet P-Y, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37(11):1207-9. 16227997 http://dx.doi.org/10.1038/ng1657
21 National Center for Biotechnology Information - NCBI [cited 2016 Sept 26]. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/7281/
Notes
Author notes
Correspondence Amir Ghorbani Aghbolaghi Department of Pathology and Laboratory Medicine - Davis Medical Center - University of California 4400 V Street - Sacramento/CA - USA 95817-1445 Phone: +1 (949) 6099916 dr.amir@me.com
Conflict of interest declaration