Image in Focus
Holoprosencephaly
Holoprosencephaly
Autopsy and Case Reports, vol. 7, no. 4, pp. 22-25, 2017
Hospital Universitário da Universidade de São Paulo
Keywords: Brain, Holoprosencephaly, Nervous system malformations
Holoprosencephaly (HPE) is a brain malformation resulting from failure of prosencephalon (the forebrain of the embryo) to divide into two distinct cerebral hemispheres. It is the most common brain malformation with an incidence of 1:250 during embryogenesis and 1:16,000 among live births.1 HPE has four subtypes: alobar holoprosencephaly, semilobar holoprosencephaly, lobar holoprosencephaly, and a middle interhemispheric fusion variant (syntelencephaly).2 Alobar holoprosencephaly is the most severe form, and as the name implies, there is no separation of the cerebral hemispheres. In semilobar holoprosencephaly, the cerebral hemispheres separate posteriorly, however are fused anteriorly. Lobar holoprosencephaly is characterized by almost complete separation of the cerebral hemispheres. Syntelencephaly results from failure of separation of posterior frontal and parietal lobes. Since both the forebrain and midface arise from the prechordal mesoderm, majority of patients with HPE also manifest craniofacial abnormalities such as microcephaly, microphthalmia, cleft lip and palate, flat nose, absent nasal bridge, and cyclopia. Multiple genetic and environmental factors are involved in the pathogenesis of HPE. Maternal diabetes mellitus is a well-known risk factor.3 Exposure to retinoic acid, diphenylhydantoin, aspirin, misoprostol, methotrexate, cholesterol-lowering agents and alcohol during pregnancy have been associated with HPE.4-9 Other environmental factors include TORCH infections during early pregnancy.1 Genetic abnormalities associated with HPE include trisomy 13, trisomy 18, and triploidy.10,11 Syndromic association of HPE includes, but is not limited to Smith-Lemli-Opitz syndrome, Genoa syndrome, Meckel-Gruber syndrome, Lambotte syndrome, Pallister-Hall syndrome, Steinfeld syndrome, caudal dysgenesis and Aicardi syndrome.12-19 Mutations in SHH, ZIC2, SIX3, and TGIF genes have been implicated in non-syndrome associated HPE.20
Central nervous system abnormalities are identified on routine prenatal imaging and etiologic diagnosis can be done by prenatal or postnatal karyotype and testing for known gene mutations. Sub classification is based on MRI findings or autopsy findings if one is requested.
Infants who survive have a myriad of clinical presentation. Some of the common physical findings include spasticity, hypotonia, choreoathetosis and dystonia. Infantile spasms and seizures are common. Feeding difficulties, gastroesophageal reflux, and malnutrition occur commonly. Other problems include temperature dysregulation and respiratory tract infections. Death usually occurs due to brainstem dysfunction or manifestation and complications of associated syndromes. Treatment is mainly supportive.
Prognosis depends upon subtype and associated syndrome.21 Those with alobar type die within days of birth.22 Around 50% with the isolated semilobar form survive beyond 1 year.22 Recurrence risk in subsequent pregnancies is high in established cases of parental carrier state and is low if the genetic abnormalities occur de novo.23,24
Figure 1 refers to gross appearance of brain in a 7-hour old female infant born to a 41-year old G1 P0 lady with limited prenatal care, past medical history of diabetes mellitus type 2 and alcohol use during first trimester of the pregnancy. On prenatal ultrasonography, the fetus had hydrocephalous and suboptimal development of cerebral and cerebellar hemispheres. Karyotyping showed normal signal pattern for chromosomes 13, 18 and 21.
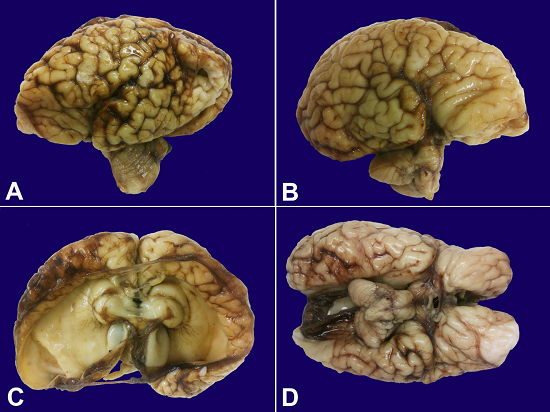
Figure 1
Macroscopic appearance of the brain depicting complex gyration without classical sulcal landmarks (A and B) and focal polymicrogyria (A), flattened cerebral hemispheres surrounding a single cystically dilated ventricle (C), fused right and left basal ganglia and diencephalic structures (C), unremarkable midbrain, cerebellum and medulla (D).
Autopsy findings included fetal macrosomia; craniofacial dysmorphogenesis to include hypertelorism, low set ears, cleft palate, absent nasal bridge and bossing of forehead; biventricular cardiomegaly, muscular ventricular septal defect and imperforated anus.
The detailed brain examination revealed flattened frontal, temporal and occipital lobes with a rudimentary C-shaped interhemispheric fissure. The surface of the cystically dilated forebrain displayed complex gyration; however, without classical sulcal landmarks (Figure 1A and 1B). There were focal polymicrogyria patches (Figure 1A). Cortical pallium surrounded a single large cystic cavity (telencephalic vesicle) in which lateral ventricles and ventricular horns could not be discerned (Figure 1 C). At the base of cystically dilated cerebrum, there were fused right and left basal ganglia and diencephalic structures (Figure 1C). There was no corpus callosum. The midbrain, cerebellum and medulla appeared unremarkable (Figure 1D).
REFERENCES
1 Solomon BD, Gropman A, Muenke M. Holoprosencephaly overview. Seattle: University of Washington; 2016 [cited 2017 August 24]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1530/
2 Barkovich AJ, Quint DJ. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol. 1993;14(2):431-40. PMid:8456724
3 Barr M Jr, Hanson JW, Currey K, et al. Holoprosencephaly in infants of diabetic mothers. J Pediatr. 1983;102(4):565-8. PMid:6834191 http://dx.doi.org/10.1016/S0022-3476(83)80185-1.
4 Edison RJ, Muenke M. Gestational exposure to lovastatin followed by cardiac malformation misclassified as holoprosencephaly. N Engl J Med. 2005;352(26):2759. 15987932 http://dx.doi.org/10.1056/NEJM200506303522622
5 Su PH, Chen JY, Lee IC, Ng YY, Hu JM, Chen SJ. Pfeiffer-like syndrome with holoprosencephaly: a newborn with maternal smoking and alcohol exposure. Pediatr Neonatol. 2009;50(5):234-8. 19856868 http://dx.doi.org/10.1016/S1875-9572(09)60069-3.
6 Kotzot D, Weigl J, Huk W, Rott HD. Hydantoin syndrome with holoprosencephaly: a possible rare teratogenic effect. Teratology. 1993;48(1):15-9. 8351644 http://dx.doi.org/10.1002/tera.1420480105
7 Orioli IM, Castilla EE. Epidemiological assessment of misoprostol teratogenicity. BJOG. 2000;107(4):519-23. 10759272 http://dx.doi.org/10.1111/j.1471-0528.2000.tb13272.x.
8 Miller EA, Rasmussen SA, Siega-Riz AM, Frías JL, Honein MA. Risk factors for non-syndromic holoprosencephaly in the National Birth Defects Prevention Study. Am J Med Genet C Semin Med Genet. 2010;154C(1):62-72. 20104597 http://dx.doi.org/10.1002/ajmg.c.30244
9 Corona-Rivera JR, Rea-Rosas A, Santana-Ramírez A, Acosta-León J, Hernández-Rocha J, Miguel-Jiménez K. Holoprosencephaly and genitourinary anomalies in fetal methotrexate syndrome. Am J Med Genet A. 2010;152A(7):1741-6. 20578136 http://dx.doi.org/10.1002/ajmg.a.33496
10 Papp C, Beke A, Ban Z, Szigeti Z, Toth-Pal E, Papp Z. Prenatal diagnosis of trisomy 13: analysis of 28 cases. J Ultrasound Med. 2006;25(4):429-35. 16567430 http://dx.doi.org/10.7863/jum.2006.25.4.429
11 Solomon BD, Rosenbaum KN, Meck JM, Muenke M. Holoprosencephaly due to numeric chromosome abnormalities. Am J Med Genet C Semin Med Genet. 2010;154C(1):146-8. 20104610 http://dx.doi.org/10.1002/ajmg.c.30232
12 Nöthen MM, Knöpfle G, Födisch HJ, Zerres K. Steinfeld syndrome: report of a second family and further delineation of a rare autosomal dominant disorder. Am J Med Genet. 1993;46(4):467-70. 8357025 http://dx.doi.org/10.1002/ajmg.1320460426
13 Cunniff C, Kratz LE, Moser A, Natowicz MR, Kelley RI. Clinical and biochemical spectrum of patients with RSH/Smith-Lemli-Opitz syndrome and abnormal cholesterol metabolism. Am J Med Genet. 1997;68(3):263-9. 9024557 http://dx.doi.org/10.1002/(SICI)1096-8628(19970131)68:3<263::AID-AJMG4>3.0.CO;2-N.
14 Balci S, Teksen F, Dökmeci F, et al. Prenatal diagnosis of Meckel-Gruber syndrome and Dandy-Walker malformation in four consecutive affected siblings, with the fourth one being diagnosed prenatally at 22 weeks of gestation. Turk J Pediatr. 2004;46(3):283-8. PMid:15503488
15 Verloes A, Dodinval P, Beco L, Bonnivert J, Lambotte C. Lambotte syndrome: microcephaly, holoprosencephaly, intrauterine growth retardation, facial anomalies, and early lethality--a new sublethal multiple congenital anomaly/mental retardation syndrome in four sibs. Am J Med Genet. 1990;37(1):119-23. 2240028 http://dx.doi.org/10.1002/ajmg.1320370128
16 Sills IN, Rapaport R, Desposito F, Lieber C. Familial Pallister-Hall syndrome: three affected offspring. Am J Med Genet. 1994;52(2):251. 7802025 http://dx.doi.org/10.1002/ajmg.1320520231
17 Sato N, Matsuishi T, Utsunomiya H, et al. Aicardi syndrome with holoprosencephaly and cleft lip and palate. Pediatr Neurol. 1987;3(2):114-6. 3508052 http://dx.doi.org/10.1016/0887-8994(87)90039-7.
18 Martínez-Frías ML, Bermejo E, García A, Galán E, Prieto L. Holoprosencephaly associated with caudal dysgenesis: a clinical-epidemiological analysis. Am J Med Genet. 1994;53(1):46-51. 7802035 http://dx.doi.org/10.1002/ajmg.1320530110
19 Camera G, Lituania M, Cohen MM Jr. Holoprosencephaly and primary craniosynostosis: the Genoa syndrome. Am J Med Genet. 1993;47(8):1161-5. 8291548 http://dx.doi.org/10.1002/ajmg.1320470806
20 Mercier S, Dubourg C, Garcelon N, et al. New findings for phenotype-genotype correlations in a large European series of holoprosencephaly cases. J Med Genet. 2011;48(11):752-60. 21940735 http://dx.doi.org/10.1136/jmedgenet-2011-100339
21 Olsen CL, Hughes JP, Youngblood LG, Sharpe-Stimac M. Epidemiology of holoprosencephaly and phenotypic characteristics of affected children: New York State, 1984-1989. Am J Med Genet. 1997;73(2):217-26. 9409876 http://dx.doi.org/10.1002/(SICI)1096-8628(19971212)73:2<217::AID-AJMG20>3.0.CO;2-S.
22 Bullen PJ, Rankin JM, Robson SC. Investigation of the epidemiology and prenatal diagnosis of holoprosencephaly in the North of England. Am J Obstet Gynecol. 2001;184(6):1256-62. 11349198 http://dx.doi.org/10.1067/mob.2001.111071
23 Mercier S, Dubourg C, Belleguic M, et al. Genetic counseling and “molecular” prenatal diagnosis of holoprosencephaly (HPE). Am J Med Genet C Semin Med Genet. 2010;154C(1):191-6. 20104616 http://dx.doi.org/10.1002/ajmg.c.30246
24 Nanni L, Ming JE, Bocian M, et al. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. 1999;8(13):2479-88. 10556296 http://dx.doi.org/10.1093/hmg/8.13.2479
Notes
Author notes
Correspondence Ameer Hamza, MD. Department of Pathology, St John Hospital and Medical Center 22101 Moross Road - Detroit/MI - USA >48236 Phone: +1 (313) 613-7511 / Fax: 313-343-8318 ameerhamza7@hotmail.com
Conflict of interest declaration