Article / Autopsy Case Report
Intravascular large B-cell lymphoma with multi-organ failure presenting as a pancreatic mass: a case with atypical presentation and definite diagnosis postmortem
Intravascular large B-cell lymphoma with multi-organ failure presenting as a pancreatic mass: a case with atypical presentation and definite diagnosis postmortem
Autopsy and Case Reports, vol. 7, no. 4, pp. 30-36, 2017
Hospital Universitário da Universidade de São Paulo
Received: 30 June 2017
Accepted: 20 September 2017
ABSTRACT: Intravascular large B-cell lymphoma (IVLBCL) is a very rare extra nodal lymphoma that tends to proliferate within small blood vessels, particularly capillaries and postcapillary venules while sparing the organ parenchyma. The cause of its affinity for the vascular bed remains unknown. Because of its rarity and unremarkable clinical presentation, a timely diagnosis of IVLBCL is very challenging. Here, we describe a case of IVLBCL presenting as pancreatic mass that was ultimately diagnosed at autopsy. A 71-year-old Caucasian female presented with a 3-month history of fatigue, abdominal pain, and weight loss. She was referred to the emergency room with a new diagnosis of portal vein thrombosis and lactic acidosis. During her hospital course she was found to have a 1.9 × 1.8 cm lesion in the pancreatic tail on imaging; The cytologic specimen on the mass showed a high-grade lymphoma. A bone marrow biopsy showed no involvement. The patient’s condition rapidly deteriorated and she, later, died due to multi-organ failure. An autopsy revealed diffuse intravascular invasion in multiple organs by the lymphoma cells. Based on our literature review—and to the best of our knowledge—there are virtually no reports describing the presentation of this lymphoma with a discernible tissue mass and associated multi-organ failure. The immunophenotypic studies performed revealed de novo CD5+ intravascular large B-cell lymphoma, which is known to be aggressive with very poor prognosis. Although it is a very rare lymphoma, it should be considered as a potential cause of multi-organ failure when no other cause has been identified. A prompt tissue diagnosis, appropriate high-dose chemotherapy and stem cell transplantation remain the only viable alternative to achieve some kind of remission.
Keywords: Pancreatic Neoplasm, Lymphoma, B-cell, Multiple Organ Failure, Autopsy.
CASE REPORT
A 71-year-old Caucasian female had a past medical history of arthritis, central serous retinopathy, remote deep vein thrombosis (not on anticoagulation), hyperlipidemia and osteoporosis. She presented with a 3-month history of fatigue, abdominal pain and loss of appetite. She was referred to the emergency room after an outpatient work-up for gradual progression of fatigue and anemia. She had a hemoglobin of 10.7 g/dL (reference value [RV]: 13.7-17.5 g/dL), mild thrombocytopenia and abnormal liver function tests. During her hospital admission, she was afebrile with the following vital signs: pulse 110 beats/minute, blood pressure 115/80 mmHg, and O2 saturation of 95% on room air. Her notable lab values were Na+ 128 mmol/L (RV: 134-146 mmol/L), AST 110 U/L (RV: 15-56 U/L), ALT 73 U/L (RV: 11-50 U/L), GGTP 94 U/L (RV: 3.0-28.7 IU/L) and albumin 2.6 gm/dL (RV: 3.5-5.0 gm/dL). The abdominal computed tomography suggested hepatic and portal vein thrombosis, with suspicion of hepatitis, pancreatitis, and colitis. On magnetic resonance imaging, right hepatic vein thrombosis and a 1.9 × 1.8 cm lesion in the pancreatic tail was noted; based on the imaging studies, a pancreatic carcinoma or a pancreatic neuro-endocrine tumor was suspected. A CT-guided needle core biopsy on the pancreatic tail mass showed multiple foci of large frankly neoplastic cells infiltrating normal-appearing pancreatic lobules featuring scant cytoplasm, nuclei with regular contour and prominent nucleoli; mitotic figures and numerous apoptotic bodies were noted. Extensive immunohistochemical work-up was unrevealing and proliferation index by Ki-67 was high (>95%). Ultimately, the patient was given a diagnosis of diffuse large B-cell lymphoma (DLBCL) on the concomitant cytology specimen, which was the actual diagnostic material. The cell block showed highly atypical large cells intermixed with pancreatic acini. Those cells were positive for CD20, CD79a, and CD45, and focally PAX-5 (Figure 1). Stain for MUM-1 was non-contributory, and a further sub-classification could not be performed. Subsequent bone marrow aspirate and biopsy showed no involvement.
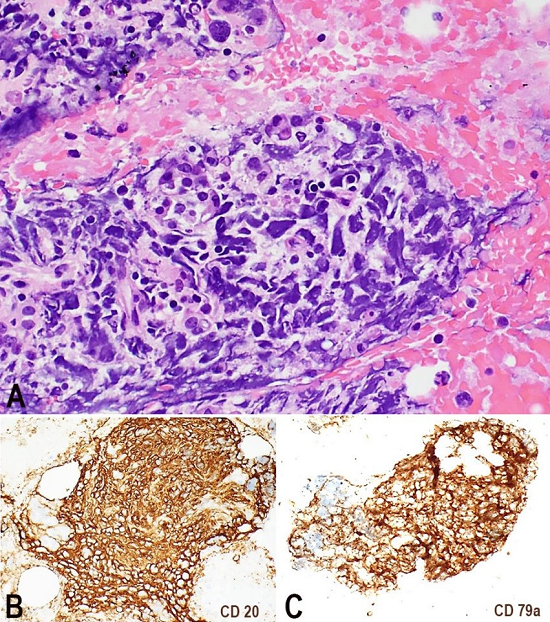
Figure 1
Photomicrography of the cell block. A - Highly atypical large cells intermixed with pancreatic acini (H&E, 20X). Those cells were positive for CD20 and CD79a in B and C, respectively (10X).
Her hospital course was complicated by Weisella confusa and Enterococcus faecalis bacteremia and she was treated with piperacillin-tazobactam (ZosynR). She was noted to have up-trending creatinine (0.7 mg/dL to 1.4 mg/dL to 1.7 mg/dL to 2.1 mg/dL; (RV: 0.6-1.0 mg/dL) and elevated lactate dehydrogenase (LDH) (9.1 U/L to 3487 U/L to 8097 U/L to 5686 U/L to 4328 U/L to 4636 U/L; (RV:140-280 U/L) of unclear etiology reminiscent of tumor lysis albeit disproportionate to lymphomatous involvement that at the time was thought to be restricted to the pancreas. Her diastolic blood pressure was intermittently fluctuating from 50 to 90 mmHg and she had up-trending liver function tests (AST/ALT: 50/47 U/L to 108/69 U/L to 410/155 U/L to 172/101 U/L), all of which were refractory to intravenous fluid. Her antibiotic regimen was broadened to vancomycin and meropenem and later she was transferred to the intensive care unit (ICU). With septic shock in mind, she was started on metronidazole (FlagylR) and oral vancomycin for Clostridium difficile colitis, empirically. Fluids and a norepinephrine drip did not improve her lactic acidosis. She was intubated due to worsening respiratory status; antibiotics were again broadened to vancomycin, meropenem, amikacin, micafungin, oral vancomycin, and metronidazole. Stress dose steroids were also given. She subsequently developed disseminated intravascular coagulopathy, pulmonary emboli and rapidly evolved into irreversible multi-organ failure ultimately resulting in the patient’s demise at day 13 after admission. There was an informed consent for the autopsy, which was performed in accordance with the institution’s ethics guidelines.
AUTOPSY FINDINGS
At autopsy, the patient was found to have a pancreatic mass (1.9 × 1.8) cm (Figure 2) composed of sheets of large lymphoma cells. Similar lymphoma cells were also seen within vessels (mainly in small vessels, capillaries, and venules) in nearly all the examined organs (lungs, liver, kidneys, thyroid, pancreas, ovary, uterus, bladder, leptomeninges, brain parenchyma, brain stem, and spinal cord dura). Larger organizing occlusive thromboemboli with predominant neoplastic cell components were identified in the left pulmonary artery (15.5 cm), and the segmental right upper and lower lobe pulmonary arteries, the right atrial thrombus (2.8 cm), and the intrahepatic right portal vein (6 cm). The pancreas showed a mass-forming 2 cm lesion composed of dysplastic lymphocytes, and the pancreas had multiple nodules of extravasated dysplastic cells (1.5 cm each).
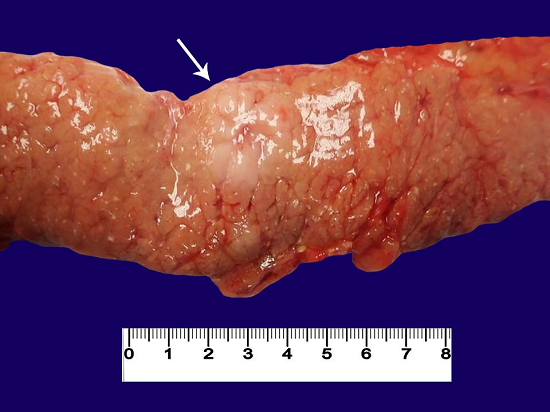
Figure 2
Post-mortem gross image of the pancreatic mass (arrow).
Pink-tan, friable, large (up to 2 cm) paraesophageal and perihepatic lymph nodes were identified at autopsy and sampled. Histopathologic examination showed hemorrhagic lymph nodes with complete effacement by large dysplastic lymphocytes and necrosis.
The spleen weighted 350 gm and had an opaque purple-gray capsule. The parenchyma was dark-red-purple, congested, and grossly without distinct masses or nodules. On histopathologic evaluation, the white pulp was effaced with diffuse sinusoidal infiltration by large pleomorphic lymphocytes.
Grossly, the liver (1520 gm) parenchyma had a yellow-brown nutmeg appearance and was diffusely infiltrated by small firm white nodules. The central liver was soft and brown, corresponding to histopathologic necrosis. There was a 6 cm occlusive thrombus in the right intrahepatic portal vein and a 2 cm occlusive thrombus in a branch of the left intrahepatic portal vein with a surrounding area (3.0 × 1.5 × 1.0 cm) that was firm, ill-defined, white, and infiltrative, which corresponded to extravasated neoplastic cells on histopathology. There were no signs of infection, grossly or histopathologically.
The portal vein thrombosis was related to neoplastic involvement, as the thrombus was predominantly composed of neoplastic lymphocytes. The neoplastic lymphoid cells were larger in size with large pleomorphic nuclei and irregular prominent nucleoli. Some anaplastic cells were seen with larger more atypical features. The tumor burdens were largely within the vasculature. CD20, PAX5, CD5, CD10, MUM1, CD3, and Ki-67 immunohistochemistry stains were obtained on the pancreatic mass (Figure 3) and the lung tissue (Figure 4). The malignant cells were positive for CD20 (strong), MUM1 (weak), Pax-5 (weak) and CD5, and were negative for CD3 and CD10. Ki-67 showed a high proliferative index (>90%). The Epstein-Barr encoding region in situ hybridization was negative.
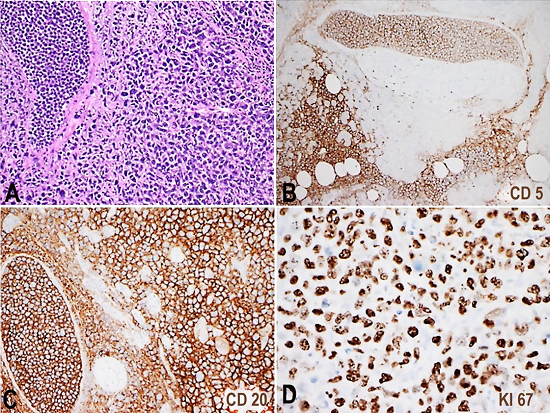
Figure 3
Photomicrography of pancreatic mass section is seen in A (H&E 10x). CD5, CD20 and Ki67(20x) immunohistochemistry stains in B, C and D, respectively, show intravascular infiltration of the lymphoma cells, which were positive for CD20 and CD5 with a high Ki67 proliferation index.
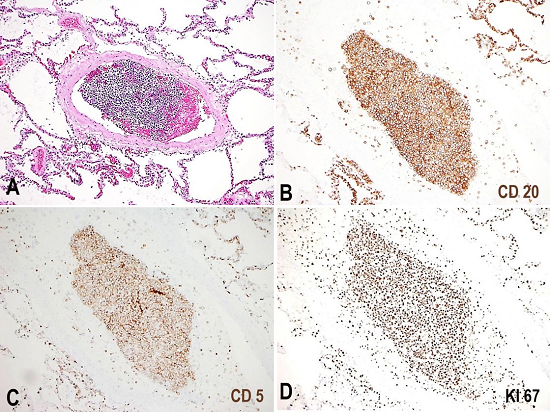
Figure 4
Photomicrography of representative lung section is seen in A (H&E 10x). CD20, CD5 and Ki67 immunohistochemistry stains in B, C and D, respectively, show intravascular infiltration by lymphoma cells positive for CD20 and CD5, with a high Ki67 proliferation index.
DISCUSSION
We report the findings on a 71-year-old woman with no significant past medical history who presented with 3-month history of fatigue, anorexia, and abdominal pain, who was found to be anemic, mildly thrombocytopenic, with abnormal liver function tests, and a ~2 cm pancreatic nodule diagnosed as DLBCL. The patient followed an inexplicably fulminant clinical course and succumbed less than 2 weeks after admission. The discrete pancreatic mass initially diagnosed as DLBCL at postmortem examination was proven to be a manifestation of widespread IVLBCL, which was an astounding discovery not previously described in the literature. A recent report has described a case of nodular goiter of the thyroid ultimately diagnosed as IVLBCL.1 However, the lymphoma cells in the thyroid were harbored exclusively intravascularly. In our case, the radiographic findings of the pancreatic mass mostly resembled pancreatic carcinoma or neuroendocrine tumor, and the initial needle core biopsy proved inconclusive after extensive immunohistochemistry targeting those entities. However, evidence of lymphomatous origin was detected at cytologic examination of concomitant fine needle aspiration; morphologic examination and immunostaining performed on the cytology cell block were diagnostic of DLBCL, which could not be further characterized due to tissue paucity. Subsequent negative clinical staging suggesting that the lymphoma was restricted to the approximately 2 cm pancreatic nodule resulted in a clinical conundrum, as the patient’s general condition rapidly deteriorated despite robust antibiotic therapy, which drew increasing skepticism among infectious disease team members regarding the alleged infectious etiology. An incomprehensible tumor lysis syndrome was contemplated but the patient’s dismal conditions cautioned against systemic chemotherapy while at the ICU. Only the postmortem examination unveiled a widely disseminated IVLBCL, which ultimately accounted for the patient’s demise. Retrospectively, we could not find evidence of an intravascular component on the cytology specimen, but it was clearly present on sections obtained at necropsy.
IVLBCL was first described in 1958 by Pfleger and Tappeiner as a very rare form of DLBCL that proliferates within small blood vessels.2 According to the 2008 WHO classification, this has been categorized as a rare type of non-Hodgkin lymphoma. The main characteristic of IVLBCL is diffuse, occlusive, intravascular, lymphomatous proliferation affecting the capillaries, small arteries, and veins.3,4 IVLBCL can involve any organ of the body, but typically the lymph nodes are spared. In 2014, Fonkem et al.5 retrospectively analyzed 740 cases of intravascular lymphoma between 1959 and 2011, among which 651 were IVLBCL. This retrospective research found that the central nervous system, bone marrow spleen, skin, and lung involvement was present in 60%, 11%, 11%, 8% and 7% of cases, respectively.
The pathogenesis of IVLBCL is unclear, and an explanation accounting for its affinity to vascular beds remains elusive. Conceivably, adhesion molecule dysfunction would partly explain why lymphoma cells remain stationary within the lumen of vessels.6 Histologically, lymphoma cells are large and mitotically active with prominent nucleoli. Histopathology remains the gold standard for diagnosis, showing the classic appearance of large malignant lymphocytes filling the small vascular lumen. Immunohistochemically, the cells express B-cell markers with an aberrant expression of CD5 and, less likely, CD10, by a portion of cases. The clinical manifestations of IVLBCL are non-specific; fever, weight loss, progressive neurologic deficits, skin findings, and sweating are the most common presentations.7 Increased serum LDH and C-reactive protein are found in most cases, accompanied by anemia and decreased platelets and a white blood cell count.
The pancreatic lesion described here with extravascular extension into the pancreatic parenchyma (as shown in Figure 3) is uncanny. We believe this represents a feature not previously described in other cases of IVLBCL. Apart from that, our case is indistinguishable from other IVLBCL, albeit we found other unusual features such as large vessel involvement, including portal vein and pulmonary arteries, and the infiltration of para-esophageal and para-aortic lymph nodes. It is to be noted that CD5+ DLBCL often presents with low-level blood involvement, as well as visceral involvement of the liver, spleen, and bone marrow, which begs the question of a possible relationship reflecting the spectrum of a disease with common lymphomatous origin.
While the diagnosis of IVLBCL is undisputable in light of the autopsy findings of extensive systemic small- and medium-sized vessel engorgement with large CD5+ lymphoma cells, the initial presentation of a discrete head of the pancreas mass that was biopsy proven CD5+ DLBCL, while the patient was still alive, represents a new clinical feature of this poorly understood lymphoma. In retrospect, this partly explains why the patient underwent tumor lysis after initial steroid therapy, which was out of proportion to the limited tumor burden she was thought to have at the time of diagnosis. This was attributed to concomitant sepsis, which the Infectious disease service deemed unlikely, although no better explanation could be hypothesized at the time.
Therefore, it is important to consider this diagnosis in the appropriate settings because patients may achieve durable remission with early diagnosis and rituximab-containing chemotherapy, which has improved the outcome of IVLBCL therapy.8-10
REFERENCES
1 Luo B, Chen JM, Liu J, et al. A case of intravascular large B cell lymphoma presenting as nodular goiter. Diagn Pathol. 2017;12(1):64. 28841887 http://dx.doi.org/10.1186/s13000-017-0656-x
2 Pfleger L., Tappeiner J. On the recognition of systematized endotheliomatosis of the cutaneous blood vessels (reticuloendotheliosis?). Hautarzt. 1959;10:359-63.
3 Ponzoni M, Ferreri AJM, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-73. 17577023 http://dx.doi.org/10.1200/JCO.2006.08.2313
4 Nakamura S, Ponzoni M, Campo E. WHO classif tumours haematop lymphoid tissues. Lyon, France: IARC; 2008. p. 252-53.
5 Fonkem E, Lok E, Robison D, Gautam S, Wong ET. The natural history of intravascular lymphomatosis. Cancer Med. 2014;3(4):1010-24. 24931821 http://dx.doi.org/10.1002/cam4.269
6 Weitten T, Rozan-Rodier S, Guiot P, Andrès E, Mootien Y. Multiorgan failure caused by intravascular lymphoma: a highly rare and malignant hemopathy mimicking multisystemic disease. South Med J. 2008;101(9):952-4. 18708972 http://dx.doi.org/10.1097/SMJ.0b013e318181291f
7 Ferreri AJM, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-83. 15461623 http://dx.doi.org/10.1111/j.1365-2141.2004.05177.x.
8 Su DW, Pasch W, Costales C, Siddiqi I, Mohrbacher A. Asian-variant intravascular large B-cell lymphoma. Proc Bayl Univ Med Cent. 2017;30(2):186-9. PMid:28405077
9 Makino K, Nakata J, Kawachi S, Hayashi T, Nakajima A, Yokoyama M. Treatment strategy for reducing the risk of rituximab-induced cytokine release syndrome in patients with intravascular large B-cell lymphoma: a case report and review of the literature. J Med Case Reports. 2013;7(1):280. 24377366 http://dx.doi.org/10.1186/1752-1947-7-280
10 Tsuyama N, Ennishi D, Yokoyama M, et al. Clinical and prognostic significance of aberrant T-cell marker expression in 225 cases of de novo diffuse large B-cell lymphoma and 276 cases of other B-cell lymphomas. Oncotarget. 2017;8(20):33487-500. PMid:28380441
Notes
Author notes
Correspondence Arnaldo A. Arbini Division of Hematopathology - Department of Pathology - NYU Langone Medical Center 240 East 38th Street, 22nd Floor - New York NY 10016 Phone: (1) 212-263-5967 arnaldo.arbini@nyumc.org
Conflict of interest declaration