Article / Clinical Case Report
Unknown primary large-cell neuroendocrine tumor
Unknown primary large-cell neuroendocrine tumor
Autopsy and Case Reports, vol. 8, no. 2, e2018025, 2018
Hospital Universitário da Universidade de São Paulo
Received: 04 February 2018
Accepted: 23 May 2018
Abstract: Large-cell neuroendocrine tumors (NETs) are poorly differentiated malignancies of rare incidence and aggressive nature. NETs mostly arise in the lung followed by the gastrointestinal tract, although they are potentially ubiquitous throughout the body. Primary unknown NET has a worse prognosis and shorter survival comparing with other NETs, with limited available data in the literature concerning this subgroup. The authors report the case of large-cell NET with supraclavicular lymph node presentation. Total excisional biopsy revealed an enlarged adenopathy 18 × 15 × 10 mm, which was extensively infiltrated by a solid malignant neoplasm composed of large cells with granular chromatin, nuclear pseudo-inclusions, high mitotic index, and focal necrosis, with a Ki 67 index 25-30% and positive immunohistochemical study for the expression of cytokeratin 8/18, chromogranin, synaptophysin, and thyroid transcriptional factor-1 (TTF-1). There was no evidence of primary location apart from two infracentimetric lung lesions that could not be accessed for biopsy and were negative at both somatostatin receptor scintigraphy and positron emission tomography. The NET relapsed with three mediastinal masses, so the patient was started on chemotherapy with carboplatin and etoposide with initial total response. Early progression showed no response to further chemotherapy regimens (temozolomide, oral etoposide); therefore, the patient was treated with local radiotherapy. This patient has an atypical long survival (54 months) compared to the literature data. In fact, there are few long-term survivors of large-cell NET and they are all related to complete surgical resection.
Keywords: Carcinoma, Neuroendocrine, Neoplasms, Unknown Primary, Neuroendocrine Tumors.
INTRODUCTION
Neuroendocrine tumors (NETs) constitute a heterogeneous group of malignancies. These rare neoplasms emerge yearly with four to five new cases/100,000 inhabitants in the United States, according to the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database between 2000 and 2004.1 The recent increasing incidence is partially justified by advances in diagnostic methods, such as immunohistochemistry and imaging.
NETs are grouped into well-differentiated tumors or poorly-differentiated carcinomas, based on the morphology and markers of proliferation (including Ki-67 and mitotic index), with distinct clinical behavior and prognosis.2-4 Poorly differentiated NETs (including both small and large cells), tend to be aggressive and grow rapidly. In contrast, well-differentiated NETs are indolent with a more favorable prognosis.5
The majority of NETs arise in the lung, followed by the gastrointestinal tract (most commonly in the lower digestive tract). Nevertheless, NETs are ubiquitous throughout the body.6-9 The unknown primary NET emerges from an occult or clinically undetectable primary site in several possible locations, such as bronchus, pancreas, gastric, colon, rectum and other sites.5 This subset of patients is the one with worse prognosis and shorter survival.6
CASE REPORT
A 75-year-old female, non-smoker, presented with a painless 16 mm adenopathy at the right supraclavicular area, with no other relevant findings at physical examination and laboratory tests. Cytology from a fine-needle biopsy revealed non-small-cell carcinoma with neuroendocrine differentiation ( Figure 1 ).
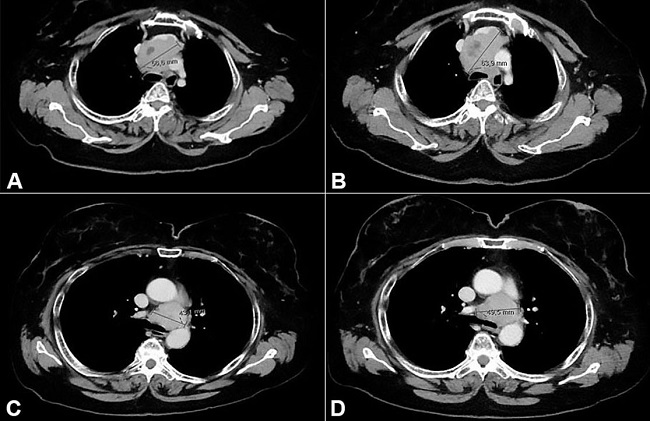
Figure 1
Fine-needle aspiration of the lymph node. A and B show the presence of large cells with anisokaryosis and pseudoinclusions (H&E 200X and 400X respectively); C and D show positivity for CD56 and TTF-1 immunostainings respectively (400X each).
The neck-chest-abdominal-pelvic computed tomography (CT) scan showed a unilateral right supraclavicular adenopathy measuring 16 mm in short axis, with no other suspicious lymph node involvement. Only two infracentimetric lung lesions (largest 4 mm) at the right inferior lobe were visible, with no other suspicious lesions/masses. Somatostatin receptor scintigraphy (SSRS) was negative, and no other relevant findings in further investigations was found (extensive laboratory workup along with upper and lower endoscopies and gynecological evaluation).
A total excisional biopsy was performed, and pathologic examination revealed an enlarged supraclavicular lymph node (18 × 15 × 10 mm), which was extensively infiltrated by a solid malignant neoplasm composed of large cells with granular chromatin, nuclear pseudo-inclusions, high mitotic index (˃20 mitotic figures per 10 high-power field), and focal necrosis. Immunohistochemical study was positive for cytokeratin 8/18, chromogranin, synaptophysin, and TTF1, in the absence of calcitonin, thyroglobulin, estrogen and progesterone receptors, mammaglobin, and S100 protein expression. Ki 67 index was 25-30%. This suggested metastatic lymph node involvement from non-small neuroendocrine carcinoma of unknown origin (possibly pulmonary) ( Figure 2 ).
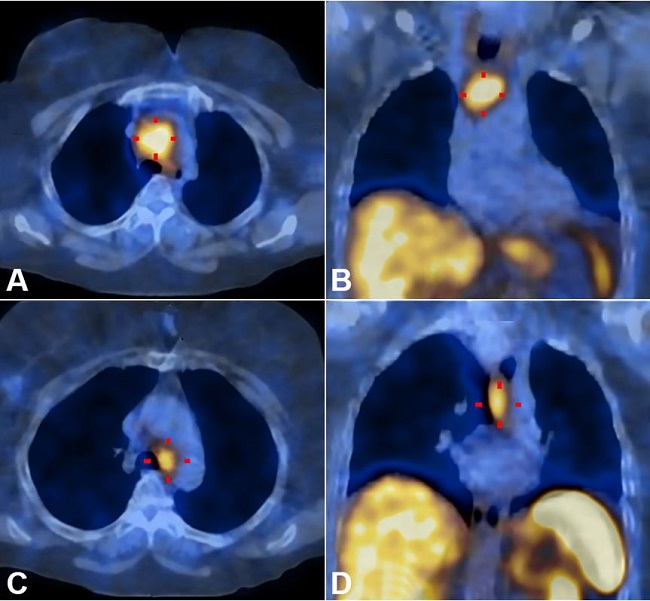
Figure 2
Histologic section of the tumor. A and B show the lymph node totally infiltrated by a solid neoplasm. Note the presence of neoplasia beyond the node’s capsule within the adjacent soft tissue (H&E 40X). The neoplastic cells show an epithelioid phenotype with overlapping features to those of the cytological sample; C, D, E and F show immunoexpression of CK8/18, chromogranin, TTF1 and synaptophysin respectively (400X).
A CT body scan re-staging showed one persistent infracentimetric lesion at the right inferior lobe of the lung, which could not be accessed for biopsy. Functional imaging with 18F-fluorodeoxyglucose positron emission tomography (18F-FDG PET) scan demonstrated three foci of intense uptake in the mediastinal lymph nodes: particularly in the projection of stations 3a (pre-vascular), 4L (left inferior para-tracheal), and 10L (left hilar), measuring 25-27 mm. Serum chromogranin A was normal.
The patient was treated with carboplatin AUC4 (day 1) and etoposide 80 mg/m2 (days 1 and 2) every 21 days. Adjustments to the standard chemotherapy protocol (carboplatin instead of cisplatin, dose reduction and 2 days’ etoposide) were prescribed considering patient’s age and comorbidities. Treatment was reasonably tolerated, and the patient presented asthenia grade 2 and total alopecia. After 6 cycles, the total metabolic response was achieved with an entirely negative 18F-FDG PET scan. Chemotherapy was then withdrawn and the oncologic surveillance was maintained.
Two months later, the patient start experiencing persistent dry cough, dysphonia, fatigue, and exertional dyspnea. Reassessment with an 18F-FDG PET scan suggested thoracic lymph node relapse with moderate 18F-FDG uptake; the CT chest scan confirmed disease progression of two mediastinal masses of 43 mm in station 3a and 33 mm in station 4L. At this stage, SSRS revealed high uptake in both mediastinal masses ( Figure 3 ).
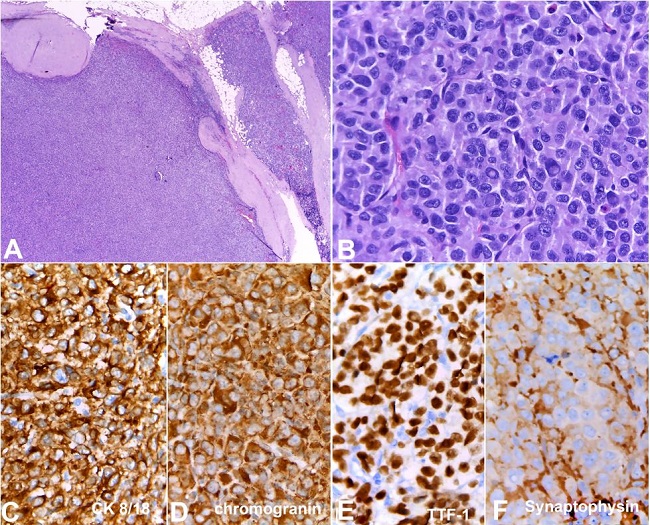
Figure 3
Somatostatin receptor scintigraphy revealed a high uptake in two mediastinal masses, consistent with disease relapse. A and B show axial and coronal planes of mediastinal mass in 3a station respectively; C and D show axial and coronal planes of mediastinal mass in 4L station respectively.
The patient was treated with lanreotide 120 mg subcutaneously every 28 days, with partial clinical improvement except for a persistent cough. After the sixth cycle, a CT body scan showed indolent disease progression with slow mediastinal mass enlargement and new confluent lymph nodes. Also, there was a slow rising of serum neuron-specific enolase (NSE). Since the disease was presenting an atypical slow-progressing course, a new biopsy was performed to guide the better therapeutic option. Therefore, the patient underwent an endobronchial ultrasound-guided transbronchial fine-needle aspiration biopsy, which confirmed the neuroendocrine carcinoma with similar morphologic characteristics of the initial biopsy.
The patient was submitted to an off-label chemotherapy with temozolomide 150 mg/m2, days 1-5, every 28 days. However, an early disease progression was verified after two cycles, with enlargement of the mediastinal masses.
The patient was treated with standard chemotherapy with oral etoposide 150 mg/m2 (days 1-3), every 21 days. No clinical nor radiologic improvement was achieved after the third cycle of treatment, with a persistent cough (non-responsive to medication) and enlargement of both mediastinal masses, although not meeting RECIST 1.1 criteria for progressive disease ( Figure 4 ).
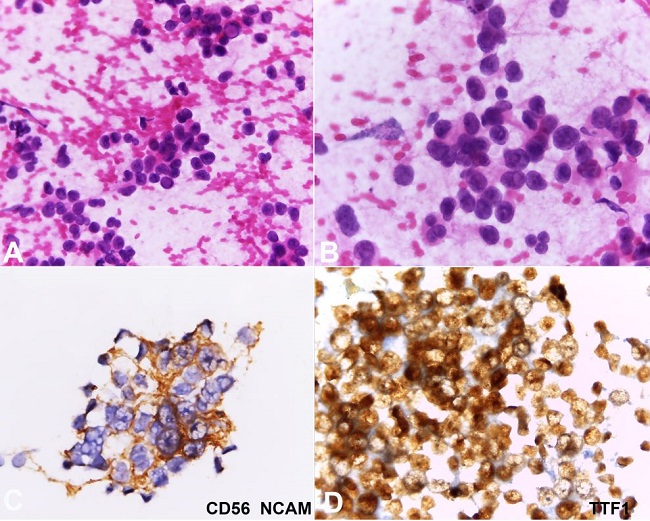
Figure 4
Computed tomography scan showing mediastinal masses enlargement during chemotherapy. A and B show axial planes of mediastinal mass in 3a station before and after oral etoposide respectively; C and D show axial planes of mediastinal mass in 4L station before and after of oral etoposide respectively.
Currently, our patient is receiving local treatment with external radiotherapy by 3D conformal technique, with 6 MV photon energy at a 2.5 Gy daily dose, over 16 planned fractions up to a 40 Gy total dose aim. The patient preserves reasonable performance status (punctuating 70% at Karnofsky scale and 1 on the Eastern Co-operative Oncology Group scale).
DISCUSSION
Suspicion remains in the face of poor clinical evidence for the primary location of our patient’s large-cell NET, which presented as supraclavicular adenopathy and later relapsed as mediastinal masses. Based on epidemiological evidence, we suspect the primary site was likely mediastinal or pulmonary.
In the literature, a similar case is reported as large-cell NET, which initially presented as an axillary lymph node, but the pulmonary origin was verified.10 In fact, poorly differentiated large-cell NET cases are reported to involve multiple metastatic sites (including liver, bone, mandibular, skin, mediastinum, and retroperitoneum lymph nodes), but a primary origin is always confirmed.11-15 There are no cases reported as truthfully primary unknown large-cell NET.
Despite the diverse presentation, large-cell NET has biological aggressive behavior, enhanced chemotherapy sensitivity and short survival.5 Gender (female) and age (< 80 years) were the favorable survival outcome predictors found in our patient. In contrast, the primary unknown site and the poorly differentiated grade were negative predictors for a better outcome.16
How to identify predictors of response and survival is still an unsolved question that some studies are engaged to answer.17,18 A relevant predictor of response to treatment is the Ki 67 index, and the cut-off value for a better therapeutic response is 55%. Another powerful predictor of survival is the performance status, so patients with good performance status had a lower percentage of immediate disease progression and a higher percentage of longer survival.17 Similarly, high values of SUVmax in the 18F-FDG PET may be a predictor of response to treatment or survival in poorly differentiated NET, although not fully clarified.
In the case presented herein, the first-line treatment choice was platinum-based chemotherapy with dose adjustments, considering the patient’s age and comorbidities.19-21 Despite a total response, the patient’s disease-free survival (5 months) was shorter when compared to series described in the literature regarding gastroenteropancreatic poorly differentiated NETs. In 1991, Moertel et al.19 reported 11 months of the median interval to progression in 18 patients with anaplastic NETs (entity analogous to the current extrapulmonary poorly differentiated NETs) treated with cisplatin 45 mg/m2 on day 1 and etoposide 130 mg/m2 for 3 days. Later, Mitry et al.20 reported median progression-free survival of 8.9 months in 41 patients with extrapulmonary poorly differentiated NET treated with cisplatin 100 mg/m2 on day 1, and etoposide 100 mg/m2 for 3 days. Most recent series and studies tend to be exclusively about NET of gastrointestinal origin.
After the consensual first-line therapy, there is no established second-line for poorly differentiated NET based on treatment guidelines.22,23 The choice of a somatostatin analogue with proven antiproliferative activity as second-line therapy was based on the new result of SSRS (positive at relapse) in a patient who was not willing to undergo further chemotherapy regimens. Lanreotide succeeded in controlling the symptoms; however, disease-free survival was shorter than that described in the literature.24 After progression, well-tolerated and expected effective chemotherapy regimens were chosen: temozolomide and oral etoposide. There are few case reports in the literature that suggest the effectiveness of temozolomide, and it is recommended by experts on treatment guidelines.22,25,26 Oral etoposide efficacy data remain in studies with intravenous etoposide and it was inferred in neuroendocrine treatment for this patient with few available therapy options.
The contrast is noticeable between the clinical behavior at disease presentation and at recurrence and progression in this particular case. Initially, the disease presented with aggressive behavior: high uptake of 18F-FDG PET, negative SSR, responsive to platinum-based chemotherapy and short progression-free survival. Later at relapse and subsequent progressions, the disease appeared to be more indolent with the novel uptake of SSR; it progressed slowly and there was no significant response to further treatments. This evolution may be due to tumor heterogeneity with different clonal cell selection and survival induced by the treatment.
Our patient’s overall survival has been unexpectedly long (54 months to date) in contrast with the literature. According to the recent SEER database analysis, median survival for high-grade NETs is 2.5 months for cases of unknown primary origin; 7.6 months for pulmonary and 14.5 months for extrapulmonary.6 Long-term survivors of large-cell NET are few, and all cases are related to complete surgical resection.27,28 Nevertheless, multimodal treatment, including surgery, chemotherapy and radiation therapy, can also achieve a good response and lead to long survival, even in inoperable cases.29
We believe that multimodal treatment has been beneficial for our patient. Host immunity factors also may have contributed to our patient’s atypically long survival, as well as other unclear disease’s features that an autopsy may help to clarify at the proper time.
The patient’s approval to report the clinical case was given as signed informed consent.
REFERENCES
1 Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-72. http://dx.doi.org/10.1200/JCO.2007.15.4377. 18565894
2 Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. 2010;39(6):707-12. http://dx.doi.org/10.1097/MPA.0b013e3181ec124e. 20664470
3 Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243-60. http://dx.doi.org/10.1097/JTO.0000000000000630. 26291008
4 Soto DE, Eisbruch A. Limited-stage extrapulmonary small-cell carcinoma: outcomes after modern chemotherapy and radiotherapy. Cancer J. 2007;13(4):243-6. http://dx.doi.org/10.1097/PPO.0b013e31813ffe7c. 17762759
5 Spigel DR, Hainsworth JD, Greco FA. Neuroendocrine carcinoma of unknown primary site. Semin Oncol. 2009;36(1):52-9. http://dx.doi.org/10.1053/j.seminoncol.2008.10.003. 19179188
6 Dasari A, Mehta K, Byers LA, Sorbye H, Yao JC. Comparative study of lung and extrapulmonary poorly differentiated neuroendocrine carcinomas: a SEER database analysis of 162,983 cases. Cancer. 2018;124(4):807-15. http://dx.doi.org/10.1002/cncr.31124. 29211313
7 Bohnenberger H, Dinter H, König A, Ströbel P. Neuroendocrine tumors of the thymus and mediastinum. J Thorac Dis. 2017;9(Suppl 15):S1448-57. http://dx.doi.org/10.21037/jtd.2017.02.02. 29201448
8 Ki EY, Park JS, Lee KH, Bae SN, Hur SY. Large cell neuroendocrine carcinoma of the ovary: a case report and a brief review of the literature. World J Surg Oncol. 2014;12(1):314. http://dx.doi.org/10.1186/1477-7819-12-314. 25314924
9 Sood A, Williamson SR, Leavitt DA. Neuroendocrine tumor of the ureter: a zebra among horses. J Endourol Case Rep. 2016;2(1):204-8. http://dx.doi.org/10.1089/cren.2016.0103. 27868098
10 Terada T. Pathologic diagnosis of large cell neuroendocrine carcinoma of the lung in an axillary lymph node: a case report with immunohistochemical and molecular genetic studies. Int J Clin Exp Pathol. 2013;6(6):1177-9. 23696939
11 Greco FA, Vaughn WK, Hainsworth JD. Advanced poorly differentiated carcinoma of unknown primary site: recognition of a treatable syndrome. Ann Intern Med. 1986;104(4):547-53. http://dx.doi.org/10.7326/0003-4819-104-4-547. 3006571
12 Hainsworth JD, Johnson DH, Greco FA. Poorly differentiated neuroendocrine carcinoma of unknown primary site (a newly recognized clinicopathologic entity). Ann Intern Med. 1988;109(5):364-71. http://dx.doi.org/10.7326/0003-4819-109-5-364. 2841895
13 Wang SC, Parekh JR, Zuraek MB, et al. Identification of unknown primary tumors in patients with neuroendocrine liver metastases. Arch Surg. 2010;145(3):276-80. http://dx.doi.org/10.1001/archsurg.2010.10. 20231629
14 Yuan C, Keating B, Farricielli LA, Zhang K. Large-cell neuroendocrine carcinoma (LCNEC) without pulmonary symptoms diagnosed in a cutaneous metastasis. Am J Case Rep. 2014;15:97-102. http://dx.doi.org/10.12659/AJCR.890094. 24624250
15 Schneider KM, Martinez AY, Guglielmi M. Large cell neuroendocrine carcinoma: topic review and a unique case of metastasis to the mandible. J Maxillofac Oral Surg. 2015;14(Suppl 1):120-6. http://dx.doi.org/10.1007/s12663-012-0362-x. 25861184
16 Boyar Cetinkaya R, Aagnes B, Myklebust TÅ, Thiis-Evensen E. Survival in neuroendocrine neoplasms: a report from a large Norwegian population-based study. Int J Cancer. 2018;142(6):1139-47. http://dx.doi.org/10.1002/ijc.31137. 29082524
17 Sorbye H, Welin S, Langer SW, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol. 2013;24(1):152-60. http://dx.doi.org/10.1093/annonc/mds276. 22967994
18 Koumarianou A, Chatzellis E, Boutzios G, Tsavaris N, Kaltsas G. Current concepts in the diagnosis and management of poorly differentiated gastrointestinal neuroendocrine carcinomas. Endokrynol Pol. 2013;64(1):60-72. 23450449
19 Moertel CG, Kvols LK, O’Connell MJ, Rubin J. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin: evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer. 1991;68(2):227-32. http://dx.doi.org/10.1002/1097-0142(19910715)68:2<227::AID-CNCR2820680202>3.0.CO;2-I. 1712661
20 Mitry E, Baudin E, Ducreux M, et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br J Cancer. 1999;81(8):1351-5. http://dx.doi.org/10.1038/sj.bjc.6690325. 10604732
21 Mitry E, Rougier P. The treatment of undifferentiated neuroendocrine tumors. Crit Rev Oncol Hematol. 2001;37(1):47-51. http://dx.doi.org/10.1016/S1040-8428(00)00073-1. 11164718
22 Pavel M, Baudin E, Couvelard A, et al. ENETS consensus guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2012;95(2):157-76. http://dx.doi.org/10.1159/000335597. 22262022
23 Oberg K, Akerström G, Rindi G, Jelic S. Neuroendocrine gastroenteropancreatic tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v223-7. http://dx.doi.org/10.1093/annonc/mdq192. 20555086
24 Caplin ME, Pavel M, Ćwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-33. http://dx.doi.org/10.1056/NEJMoa1316158. 25014687
25 Berbari B, Romano A, Bhatti R et al. A case report of a poorly differentiated neuroendocrine carcinoma diagnosed in the bone marrow. Gastroenterol Hepatol Open Access. 2015;3(2):73.
26 Koumarianou A, Kaltsas G, Kulke MH, et al. Temozolomide in advanced neuroendocrine neoplasms: pharmacological and clinical aspects. Neuroendocrinology. 2015;101(4):274-88. http://dx.doi.org/10.1159/000430816. 25924937
27 Jungraithmayr W, Kayser G, Passlick B, Eggeling S. Neuroendocrine differentiation and neuroendocrine morphology at two different patterns in large-cell bronchial carcinomas: outcome after complete resection. World J Surg Oncol. 2006;4(1):61. http://dx.doi.org/10.1186/1477-7819-4-61. 16953887
28 Kamiyoshihara M, Ibe T, Igai H, et al. Roentgenological occult large-cell neuroendocrine carcinoma: report of a long-term survivor. Respir Med Case Rep. 2013;8:14-7. http://dx.doi.org/10.1016/j.rmcr.2012.12.00. 26029607
29 Shimono C, Suwa K, Sato M, et al. Large cell neuroendocrine carcinoma of the gallbladder: long survival achieved by multimodal treatment. Int J Clin Oncol. 2009;14(4):351-5. http://dx.doi.org/10.1007/s10147-008-0843-6. 19705247
Notes
Author notes
Correspondence Sara Póvoa Medical Oncology Department - Centro Hospitalar de São João Alameda Prof. Hernâni Monteiro, Porto – Portugal CEP: 4200-319 Phone: +351 914072048 povoa.sara@gmail.com
Conflict of interest declaration