Image in Focus
Received: 08 July 2018
Accepted: 26 July 2018
DOI: https://doi.org/10.4322/acr.2018.040
Keywords: Osteogenesis imperfecta, Osteogenesis, Bone diseases
Osteogenesis imperfecta (OI) is a group of rare connective tissue diseases in which the main cause is directly related to the synthesis of type I collagen.1,2 OI is characterized by increased susceptibility to fractures and bone deformities, presenting varying degrees of severity and defects in type I collagen,3 both quantitatively and qualitatively.4 In the United States, the disease is present in 1 per 10,000 births with no gender predominance.5 Type I collagen coding is performed by the genes COL1A1 and COL1A23. In OI, more than 1500 dominant mutations in these and other genes have already been identified. These mutations are responsible for altering the structure and/or the amount of type I collagen.5,6 This large number of possible mutations is responsible for the variable phenotypes. In 1979, Sillence and Rimoin6 developed a classification system for osteogenesis imperfecta, dividing it into subtypes based on the clinical characteristics and severity of the disease: OI type I, autosomal dominant inheritance with bluish sclera; OI type II, perinatal lethal form; OI type III, progressive deformation, with normal sclera; and OI type IV, autosomal dominant inheritance with normal sclera.4
The disease begins its manifestation in the neonatal period, in which it is possible to identify fractures and apparent osteoporosis from radiological images. Early intervention is essential but it is not always possible to avoid all fractures.3,4 Due to the early clinical manifestations, an intrauterine diagnosis can be made. Short, arcuate limbs associated with a series of osteochondrodysplasias may be radiologically detected. The non-mineralization of the skull or visceral herniation are incompatible with life. Laboratory tests are not effective for diagnosis, but are necessary to rule out neonatal hyperparathyroidism and hypophosphatasia.3-5 As most of the cases are related to dominant inheritance, family history should be analyzed in detail, including family members with fractures, osteoporosis, and other OI-associated features such as hernias, dislocations, brittle teeth, persistent blue sclera, death before the age of 50 and retarded motor development.6 Diagnosis is made from clinical manifestations, radiological exams, bone densitometry and markers of bone metabolism and collagen.5,6
Figure 1 refers to a female preterm infant born to a 24-year-old G2 P1 A0 woman with regular prenatal care and no relevant past medical history. Prenatal ultrasonography showed that the fetus had severe bone dysplasia, bone hypomineralization, thoracic hypoplasia, craniosynostosis, micromelia, bone tortuosities and intrauterine growth restriction.
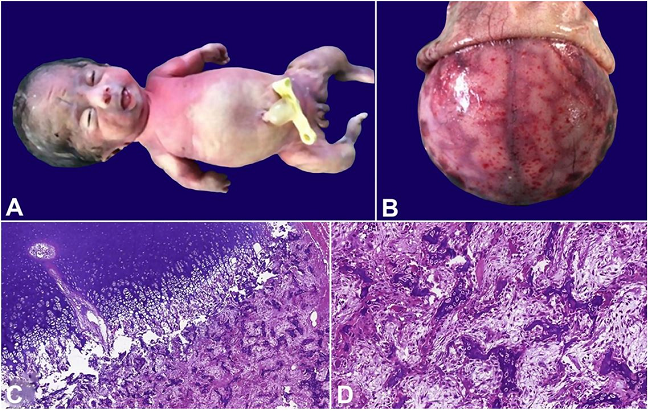
Figure 1
A - Female newborn with small thorax, a bulging abdomen and short and angled limbs; B - Examination of the cephalic segment reveals absence of the skull cap with the brain covered by a thin translucent membrane; C - Photomicrograph of the bone tissue with normal orientation of the chondrocyte forming columns, but without an efficient endochondral ossification (H&E, 5X); D - the trabecular bone shows osteopenia and disorganized bone tissue, with a reduction of the osteoid matrix (H&E, 20X).
Autopsy revealed a female newborn with a hypoplastic thorax and a bulging abdomen, presenting deformities of the limbs, which were short and angled (Figure 1A). The cranial bone was absent and the brain was covered by a thin, smooth shiny translucent membrane (Figure 1B). No other internal malformations were identified, other than pulmonary hypoplasia. The histology of the long bones revealed that the trabecular bone consisted of delicate and disorganized bone tissue with the appearance of osteopenia, presenting a reduction in the osteoid matrix. The physis and chondrocyte columns appeared normal, but there was a lack of endochondral ossification (Figure 1C and 1D). The cortical bone was thin and absent in large areas.
References
1 Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7(9):540-57. http://dx.doi.org/10.1038/nrendo.2011.81. PMid:21670757
2 Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014;164A(6):1470-81. http://dx.doi.org/10.1002/ajmg.a.36545. PMid:24715559
3 Bishop N. Characterising and treating osteogenesis imperfecta. Early Hum Dev. 2010;86(11):743-6. http://dx.doi.org/10.1016/j.earlhumdev.2010.08.002. PMid:20846798
4 Martin E, Shapiro JR. Osteogenesis imperfecta: epidemiology and pathophysiology. Curr Osteoporos Rep. 2007;5(3):91-7. http://dx.doi.org/10.1007/s11914-007-0023-z. PMid:17925189
5 Obafemi AA, Bulas DI, Troendle J, Marini JC. Popcorn calcification in osteogenesis imperfecta: incidence, progression, and molecular correlation. Am J Med Genet A. 2008;146A(21):2725-32. http://dx.doi.org/10.1002/ajmg.a.32508. PMid:18798308
6 Sillence DO, Rimoin DL. Classification of osteogenesis imperfecta. Lancet. 1978;1(8072):1041-2. http://dx.doi.org/10.1016/S0140-6736(78)90763-8. PMid:76956
Notes
Author notes
Correspondence Bruno Kusznir Vitturi Santa Casa de São Paulo - School of Medical Sciences Rua Doutor Cesário Mota Júnior, 61 - Vila Buarque - São Paulo/SP - Brazil CEP: 01221-020 Phone: +55 (11) 9757-67987 z_azul@hotmail.com
Conflict of interest declaration
