Article / Clinical Case Report
Received: 18 June 2018
Accepted: 15 August 2018
DOI: https://doi.org/10.4322/acr.2018.045
Abstract: Desmoid tumors develop from connective tissue, fasciae, and aponeuroses, and may occur in the context of familial adenomatous polyposis or may arise sporadically; also, they may be extra-abdominal, intra-abdominal, or located in the abdominal wall. These benign tumors have a great aggressiveness with a high rate of local recurrence. Familial adenomatous polyposis is an inherited condition with autosomal dominant transmission, and is characterized by the development of multiple colonic and rectal adenomatous polyps, as well as desmoid tumors. We present the case of a 54-year-old woman with germline APC gene mutation, who underwent a total colectomy, subsequently developing two large infiltrative solid intra-abdominal lesions consistent with desmoid tumors. Medical treatment with Cox-2 inhibitors was initiated without result. She was submitted to resection for intestinal obstruction, but developed local recurrence. The lesions were also unresponsive to tamoxifen, and chemotherapy was initiated with dacarbazine plus doxorubicin, switching to vinorelbine plus methotrexate, achieving a good response in all lesions after 12 months. The approach to these intra-abdominal lesions should be progressive, beginning with observation, then a medical approach with non-steroidal anti-inflammatory drugs or with an anti-hormonal agent. Afterwards, if progression is still evident, chemotherapy should be started. Surgery should be reserved for resistance to medical treatment, in palliative situations, or for extra-abdominal or abdominal wall desmoids tumors.
Keywords: Fibromatosis, Aggressive, Neoplastic Syndromes, Hereditary, Adenomatous Polyposis Coli, Drug Therapy.
INTRODUCTION
Desmoid tumors (DT) develop from connective tissue, fasciae, and aponeuroses,1,2 corresponding to a monoclonal proliferation of well-differentiated fibroblasts.3-5 This can occur in the context of familial adenomatous polyposis (FAP), or may arise sporadically; also they may be extra-abdominal, intra-abdominal, or located in the abdominal wall.5 DT is considered a benign neoplasm due to the absence of metastasis, but has a great local aggressiveness, given the infiltrative growth and the invasion of adjacent structures,6 with a high local recurrence rate, ranging from 25% to 85%.2,7
The familial form of this neoplasm may occur between 10% and 25% of FAP patients and is attributed to a germline mutation in the adenomatous polyposis coli (APC) gene.8,9 The carriers of these mutations have “850 times greater risk than the general population.”2,10 The sporadic forms of DT result from somatic mutations in the APC or the beta-catenin genes5,11 and are relatively rare, affecting approximately two to five individuals per million per year in the general population.12,13
FAP is an inherited condition “that results from the autosomal dominant transmission of a germline mutation in the APC tumor suppressor gene,” which is located on the long arm of chromosome 5.10 This pathology is characterized by the development of several (hundreds to thousands) of adenomatous polyps in the colon and rectum, with a subsequent risk of developing colorectal carcinoma.14 These patients have the risk of extra-colic manifestations like DT, upper gastrointestinal tract adenomas, osteomas, epidermoid cysts, as well as thyroid, adrenal gland, and central nervous system neoplasms.7 Of these, DT are the most frequent cause of death (after colorectal carcinoma) in patients with FAP, with mortality ranging from 18% to 31% (more than periampullary carcinomas at about 22%).15
CASE REPORT
We present the case of a 54-year-old woman with FAP in the context of germline APC mutation. She had undergone an open total colectomy 8 years before, after more than 10 years of endoscopic surveillance. Pathology revealed one site with an in situ adenocarcinoma (without positive nodes on the 68 isolated) and more than 100 adenomatous polyps with dysplasia. The patient preferred to preserve the rectum and maintain endoscopic surveillance.
During follow-up, an infiltrative solid intra-abdominal lesion in the root of the mesentery was identified in computed tomography and confirmed by magnetic resonance imaging. The hypothesis of peritoneal carcinomatosis was raised. A surgical biopsy was performed, revealing a fusiform cell tumor, which was consistent with reactive fibrosis.
At a multidisciplinary meeting, since the lesion was large, it was decided to initiate medical treatment with Cox-2 inhibitors, but no therapeutic response was observed. The disease progressed, and an intestinal obstruction developed due to a new lesion in the root of the mesentery, which measured 157 × 140 × 95 mm. Therefore, the patient underwent an en-bloc segmental enterectomy and R2 resection of the retroperitoneal tumor, with histological confirmation of a DT (Figure 1A-D). C-Kit mutations and the presence of estrogen-receptors were tested, but both markers were negative.
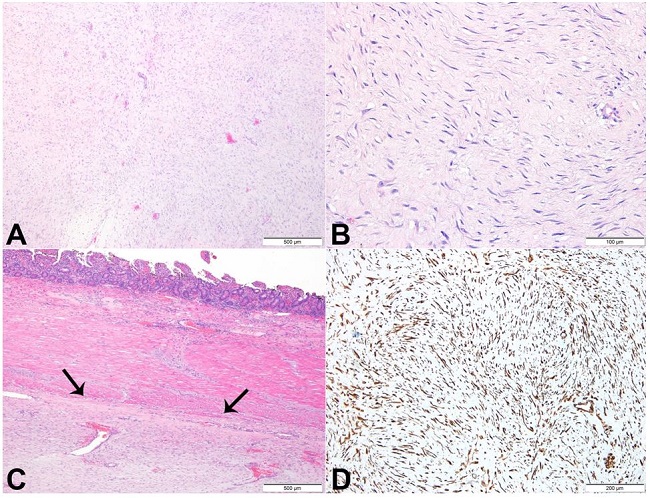
Figure 1
Photomicrograph of the retroperitoneal tumor. A - A sparse proliferation of elongated, slender, spindle-shaped cells of uniform appearance, set in a collagenous stroma (H&E); B - Cells with lack of hyperchromasia or atypia and with small, pale-staining nuclei. These cells are usually arranged in sweeping bundles (H&E); C - Lesion within the enteric wall (arrows) (H&E); D - Histochemistry showing strong expression of vimentin.
The patient was started on tamoxifen initially with 20 mg/day, increasing up to 40 mg/day, but because of a diagnosis of cholelithiasis it was decreased again to 20 mg/day. However, due to disease progression with the appearance of new lesions (Figure 2A), the chemotherapy regimen with dacarbazine (300 mg/m2) and doxorubicin (20 mg/m2)—administered at day 1, 2, and 3, and repeated every 21 days—was approved by the Institutional Ethics Committee. This regimen was further switched, after eight cycles, to vinorelbine (25 mg/m2) and methotrexate (30 mg/m2) because of the patient’s intolerance (with myelosuppression) and the progression of the pelvic lesion—albeit with the regression of all others. There was a significant response to this last protocol—administered at day 1, 8, and 15, and repeated every 28 days—with dimensional regression of all lesions (Figure 2B) and pain resolution after 14 cycles of chemotherapy.
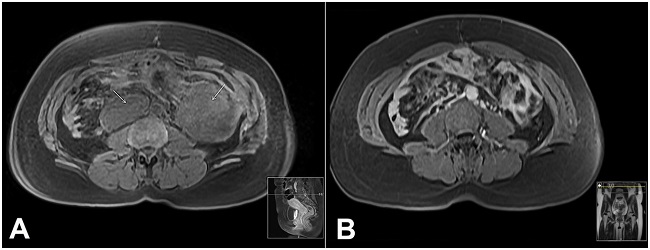
Figure 2
A - Abdominal magnetic resonance imaging (MRI) T1-weighted image, acquired in the axial plane, showing two new lesions (arrows); B - Abdominal MRI T1-weighted image, acquired in the axial plane, after chemotherapy, showing the regression of previous lesions.
DISCUSSION
DT may arise sporadically or in the context of an extra-colic manifestation of FAP, and may be intra-abdominal, located in the abdominal wall, or extra-abdominal.5,10,11 They comprise only 0.03% of all neoplasms and 3% of all soft tissue tumors. The majority of cases occur between the ages of 15 and 60 years, with a peak incidence between 25 and 35 years.16
Despite being considered benign tumors, due to the lack of metastasizing capability DT has great local aggressiveness and the potential for local recurrence.6,17 The most frequent locations are the small-bowel mesentery, followed by the abdominal wall, the extremities, and the trunk.12
The etiology of this condition is still not well understood. However, trauma or muscle strain (like surgical stress) induces a disproportionate inflammatory response, with an accumulation of fibroblasts. Another theory is that there is an endocrine factor related to neoplasm, since DT are more common in women—either when they’re pregnant or using hormonal contraceptives, and some women develop the tumor without previous trauma.17,18
Therefore, surgical trauma may be implicated as one of the factors capable of leading to the development of DT, and appears unrelated to a specific type of surgery.8 Nevertheless, some authors observed a lower incidence of DT in laparoscopic versus open procedures, so minimally invasive approaches might presumably reduce the risk of the occurrence of DT after prophylactic colectomy in patients with FAP.10,19
The mutation site on the APC gene may also interfere with the risk of development of DT and its severity. Mutations located distally to codon 1309 and especially at the 3′ end, are associated with a high risk of developing DT, and particularly in a severe form.12,20,21 Bertario et al.8 stated that, in patients with FAP, mutations located beyond codon 1309 and codon 1444 (“desmoid region”) increase 17-fold and 12-fold, respectively, the risk of DT development, compared with mutations located upstream.
In the case of intra-abdominal DT, the approach should be progressive,10 initially with observation,4 followed by medical treatment, as the first line, with non-steroidal anti-inflammatory drugs (NSAIDs; e.g. sulindac or celecoxib, with an efficacy rate close to 50%), or an anti-hormonal agent (tamoxifen or toremifene),14,22,23 even in estrogen receptor-free DT, because this therapy may still induce a response in that setting.24,25 If tumor progression persists, chemotherapy should be started.10,26 Surgery remains the final option10 in cases of resistance to medical treatment for symptomatic control, or resolution of tumor-related complications, such as intestinal obstruction, ischemia, enteric fistula, and hydronephrosis.1,27,28
However, in resectable extra-abdominal or abdominal wall DT, surgery may be an appropriate first-line treatment.29 Otherwise, a progressive approach similar to intra-abdominal DT is preferred.10
Although controversial, radiation therapy may be an alternative to surgery.30 Studies6,31 confirm the important role of such therapeutic modality in cases of unresectable abdominal wall,32 extra-abdominal DT, or after R1 resections. Either pre- or post-operatively, radiotherapy can model fibrosis, with the consequent decrease in local recurrence.14,33 In intra-abdominal DT, radiotherapy is rarely used, because of the low tumoral radiosensitivity10 and because of the risk of radiation enteritis.14
The role of chemotherapy in this setting was demonstrated by the efficacy of low doses of doxorubicin and dacarbazine, as well as the combination of methotrexate and vinblastine, in a progressive disease that did not respond to the medical therapy, which consisted in NSAIDs and anti-hormonal agents.26,34-36 Other combination therapy that demonstrated good response was cyclophosphamide plus doxorubicin,26 and doxorubicin plus tyrosine kinase inhibitors (TKI).37 For example, imatinib may have a role in the management of unresectable or difficult to resect DT.38 Sorafenib, another TKI, demonstrated a response in DT, especially in the extra-abdominal variety, and may be an option when chemotherapy fails.37
CONCLUSION
These patients should be managed by a multidisciplinary team, with individualized approaches and decisions. The strategy in intra-abdominal DT should be progressive, beginning with observation, followed by NSAIDs (e.g. sulindac), and/or an anti-hormonal agent (e.g. tamoxifen). If an unsatisfactory response persists and disease progression ensues, chemotherapy needs to be started. Surgery should be reserved for the final option, or for the resolution of tumor-related complications.
The authors declare that they have collected an informed consent from the patient to publish this article.
REFERENCES
1 Tanaka K, Toiyama Y, Okugawa Y, et al. Cytoreductive strategy for multiple intra-abdominal and abdominal wall desmoid tumors in familial adenomatous polyposis: report of three cases. Clin J Gastroenterol. 2012;5(5):361-6. http://dx.doi.org/10.1007/s12328-012-0330-5. PMid:26181076
2 Ferenc T, Sygut J, Kopczynski J, et al. Aggressive fibromatosis (desmoid tumors): definition, occurrence, pathology, diagnostic problems, clinical behavior, genetic background. Pol J Pathol. 2006;57(1):5-15. PMid:16739877
3 Colombo C, Foo WC, Whiting D, et al. FAP-related desmoid tumors: a series of 44 patients evaluated in a cancer referral center. Histol Histopathol. 2012;27(5):641-9. PMid:22419028
4 Fiore M, Rimareix F, Mariani L, et al. Desmoid-type fibromatosis: a front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol. 2009;16(9):2587-93. http://dx.doi.org/10.1245/s10434-009-0586-2. PMid:19568815
5 Leal RF, Silva PVVT, Ayrizono MLS, Fagundes JJ, Amstalden EMI, Coy CSR. Desmoid tumor in patients with familial adenomatous polyposis. Arq Gastroenterol. 2010;47(4):373-8. http://dx.doi.org/10.1590/S0004-28032010000400010. PMid:21225148
6 Pikaar A, Nortier JW, Griffioen G, Vasen HF. Desmoid tumors in patients with familial adenomatous polyposis. Ned Tijdschr Geneeskd. 2002;146(29):1355-9. PMid:12162172
7 Righetti AEM, Jacomini C, Parra RS, Almeida ALNR, Rocha JJR, Féres O. Familial adenomatous polyposis and desmoid tumors. Clinics. 2011;66(10):1839-42. http://dx.doi.org/10.1590/S1807-59322011001000027. PMid:22012061
8 Bertario L, Russo A, Sala P, et al. Genotype and phenotype factors as determinants of desmoids tumors in patients with familial adenomatous polyposis. Int J Cancer. 2001;95(2):102-7. http://dx.doi.org/10.1002/1097-0215(20010320)95:2<102::AID-IJC1018>3.0.CO;2-8. PMid:11241320
9 Friedl W, Caspari R, Sengteller M, et al. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut. 2001;48(4):515-21. http://dx.doi.org/10.1136/gut.48.4.515. PMid:11247896
10 Freitas ACR, Barbosa LER. Genetic profile, risk factors and therapeutic approach of desmoid tumors in familial adenomatous polyposis. J Coloproctol (Rio J). 2017;37(2):163-70. http://dx.doi.org/10.1016/j.jcol.2017.04.001
11 Fisher C, Thway K. Aggressive fibromatosis. Pathology. 2014;46(2):135-40. http://dx.doi.org/10.1097/PAT.0000000000000045. PMid:24378386
12 Schiessling S, Kihm M, Ganschow P, Kadmon G, Büchler MW, Kadmon M. Desmoid tumour biology in patients with familial adenomatous polyposis coli. Br J Surg. 2013;100(5):694-703. http://dx.doi.org/10.1002/bjs.9053. PMid:23334997
13 Fallen T, Wilson M, Morlan B, Lindor NM. Desmoid tumors—a characterization of patients seen at the Mayo Clinic 1976-1999. Fam Cancer. 2006;5(2):191-4. http://dx.doi.org/10.1007/s10689-005-5959-5. PMid:16736290
14 Latchford AR, Sturt NJ, Neale K, Rogers PA, Phillips RK. A 10-year review of surgery for desmoid disease associated with familial adenomatous polyposis. Br J Surg. 2006;93(10):1258-64. http://dx.doi.org/10.1002/bjs.5425. PMid:16952208
15 Seow-Choen F. The management of desmoids in patients with familial adenomatous polyposis (FAP). Acta Chir Iugosl. 2008;55(3):83-7. http://dx.doi.org/10.2298/ACI0803083S. PMid:19069698
16 Xie Y, Xie K, Gou Q, He J, Zhong L, Wang Y. Recurrent desmoid tumor of the mediastinum: a case report. Oncol Lett. 2014;8(5):2276-8. http://dx.doi.org/10.3892/ol.2014.2431. PMid:25295113
17 Nagano S, Passos R, Santana M, et al. Tumor desmoide - Uma revisão de literatura. Rev Pat Tocantins. 2015;2:2-7.
18 Okuno S. The enigma of desmoid tumors. Curr Treat Options Oncol. 2006;7(6):438-43. http://dx.doi.org/10.1007/s11864-006-0019-4. PMid:17032556
19 Vitellaro M, Sala P, Signoroni S, et al. Risk of desmoid tumours after open and laparoscopic colectomy in patients with familial adenomatous polyposis. Br J Surg. 2014;101(5):558-65. http://dx.doi.org/10.1002/bjs.9411. PMid:24493089
20 Sturt NJ, Gallagher MC, Bassett P, et al. Evidence for genetic predisposition to desmoid tumours in familial adenomatous polyposis independent of the germline APC mutation. Gut. 2004;53(12):1832-6. http://dx.doi.org/10.1136/gut.2004.042705. PMid:15542524
21 Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001;10(7):721-33. http://dx.doi.org/10.1093/hmg/10.7.721. PMid:11257105
22 Basdanis G, Papadopoulos VN, Panidis S, et al. Desmoid tumor of mesentery in familial adenomatous polyposis: a case report. Tech Coloproctol. 2010;14(Suppl 1):S61-2. http://dx.doi.org/10.1007/s10151-010-0613-0. PMid:20683751
23 Ohashi T, Shigematsu N, Kameyama K, Kubo A. Tamoxifen for recurrent desmoid tumor of the chest wall. Int J Clin Oncol. 2006;11(2):150-2. http://dx.doi.org/10.1007/s10147-005-0543-4. PMid:16622751
24 Bocale D, Rotelli MT, Cavallini A, Altomare DF. Anti-oestrogen therapy in the treatment of desmoid tumours: a systematic review. Colorectal Dis. 2011;13(12):e388-95. http://dx.doi.org/10.1111/j.1463-1318.2011.02758.x. PMid:21831172
25 Chao AS, Lai CH, Hsueh S, Chen CS, Yang YC, Soong YK. Successful treatment of recurrent pelvic desmoid tumour with tamoxifen: case report. Hum Reprod. 2000;15(2):311-3. http://dx.doi.org/10.1093/humrep/15.2.311. PMid:10655300
26 Okuno SH, Edmonson JH. Combination chemotherapy for desmoid tumors. Cancer. 2003;97(4):1134-5. http://dx.doi.org/10.1002/cncr.11189. PMid:12569616
27 Nieuwenhuis MH, Mathus-Vliegen EM, Baeten CG, et al. Evaluation of management of desmoid tumours associated with familial adenomatous polyposis in Dutch patients. Br J Cancer. 2011;104(1):37-42. http://dx.doi.org/10.1038/sj.bjc.6605997. PMid:21063417
28 Martins S, Leite J, Oliveira A, et al. Tratamento dos tumores desmoides intra-abdominais associados à Polipose Adenomatosa Familiar. Rev Port Cir. 2015;32:17-25.
29 Jung WB, Kim CW, Kim JC. Clinical characteristics adequate treatment of familial adenomatous polyposis combined with desmoid tumors. Cancer Res Treat. 2014;46(4):366-73. http://dx.doi.org/10.4143/crt.2013.185. PMid:25152189
30 Nuyttens JJ, Rust PF, Thomas CR Jr, Turrisi AT 3rd. Surgery versus radiation therapy for patients with aggressive fibromatosis or desmoid tumors. Cancer. 2000;88(7):1517-23. http://dx.doi.org/10.1002/(SICI)1097-0142(20000401)88:7<1517::AID-CNCR3>3.0.CO;2-9. PMid:10738207
31 Ballo MT, Zagars GK, Pollack A. Radiation therapy in the management of desmoid tumors. Int J Radiat Oncol Biol Phys. 1998;42(5):1007-14. http://dx.doi.org/10.1016/S0360-3016(98)00285-5. PMid:9869223
32 Turina M, Pavlik CM, Heinimann K, Behrensmeier F, Simmen HP. Recurrent desmoids determine outcome in patients with Gardner syndrome: a cohort study of three generations of an APC mutation-positive family across 30 years. Int J Colorectal Dis. 2013;28(6):865-72. http://dx.doi.org/10.1007/s00384-012-1600-x. PMid:23114473
33 Palacios-Zertuche JT, Cardona-Huerta S, Juárez-García ML, Valdés-Flores E, Muñoz-Maldonado GE. Case report: rapidly growing abdominal wall giant desmoid tumour during pregnancy. Cir Cir. 2017;85(4):339-43. http://dx.doi.org/10.1016/j.circir.2016.04.004. PMid:27318390
34 Toiyama Y, Konishi N, Inoue Y, et al. Successful treatment of ileal pouch desmoids using multimodal chemotherapy with low-dose vinblastine and methotrexate in a patient with familial adenomatous polyposis. Clin J Gastroenterol. 2009;2(3):170-4. http://dx.doi.org/10.1007/s12328-008-0055-7. PMid:26192289
35 Yamamoto H, Oshiro R, Nishimura J, et al. Low-dose dacarbazine-doxorubicin therapy against intra-abdominal desmoid tumors. Oncol Rep. 2013;29(5):1751-5. http://dx.doi.org/10.3892/or.2013.2345. PMid:23503528
36 Gega M, Yanagi H, Yoshikawa R, et al. Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol. 2006;24(1):102-5. http://dx.doi.org/10.1200/JCO.2005.02.1923. PMid:16382119
37 Gounder M, Lefkowitz R, Keohan M, et al. Activity of sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res. 2011;17(12):4082-90. http://dx.doi.org/10.1158/1078-0432.CCR-10-3322. PMid:21447727
38 Chugh R, Wathen JK, Patel SR, et al. Efficacy of imatinib in aggressive fibromatosis: results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res. 2010;16(19):4884-91. http://dx.doi.org/10.1158/1078-0432.CCR-10-1177. PMid:20724445
Notes
Author notes
Correspondence Vítor Devezas Department of Surgery - Faculty of Medicine - Centro Hospitalar de São João (CHSJ) Alameda Prof. Hernâni Monteiro - Porto - Portugal 4200-319 Phone: +351 (93) 742-7911 / Fax: + 351 (22) 502-5766 vitor.devezas7@gmail.com
Conflict of interest declaration
