Artículos originales
Análisis del genoma completo de las cepas emergentes de rotavirus G9P[4] y G8P[8] en Argentina: nueva introducción vs. reordenante de cepas locales
Complete genome analysis of emergent G9P[4] and G8P[8] rotavirus strains in Argentina: novel introduction vs. re-assortment of local strains

Análisis del genoma completo de las cepas emergentes de rotavirus G9P[4] y G8P[8] en Argentina: nueva introducción vs. reordenante de cepas locales
Bioquímica y Patología Clínica, vol. 89, núm. 1, pp. 44-58, 2025
Asociación Bioquímica Argentina
Resumen: Introducción: Un rotavirus (RVA) presenta asociaciones G-/P- comunes e inusuales. El reordenamiento entre cepas durante una coinfección constituye uno de sus mecanismos más importantes en la generación de diversidad genética. Durante el período 2017-2018, se detectaron dos asociaciones inusuales (G9P[4] y G8P[8]) por primera vez en Argentina. Se desconoce si representan nuevas introducciones o si son producto de un reordenamiento entre asociaciones comunes locales. Objetivo: Analizar las relaciones filogenéticas y evolutivas de G9P[4] y G8P[8] con respecto a las asociaciones comunes circulantes del mismo período. Materiales y métodos: Se incluyeron cepas de RVA representativas de asociaciones comunes (n=11) e inusuales (n=2), detectadas durante el período 2016-2018, en el marco de la vigilancia epidemiológica nacional. Partiendo de materia fecal, se realizó la extracción de ácidos nucleicos y se amplificaron y secuenciaron los 11 segmentos genómicos. Se determinaron las constelaciones genéticas, se construyeron árboles filogenéticos y se elaboraron matrices de distancia en los niveles nucleotídico y aminoacídico. Resultados: Las constelaciones obtenidas fueron G9P[4]-I2-R2-C2-M2-A2-N2-T2-E6-H2 y G8P[8]-I2-R2-C2-M2-A2-N2-T2-E2-H2. La G8P[8] se relacionó en 10 segmentos con una G3P[8] que circuló en 2016. G9P[4] se relacionó en 6 genes con una G2P[4] que circuló en 2016, en 2 genes, con una G3P[8] del 2016 y en un gen, con una_ G2P[4] del 2017. . Sin embargo, no se observaron fenómenos de reordenamiento en ninguno de los 11 segmentos. Conclusiones: Las cepas emergentes G9P[4] y G8P[8], detectadas en Argentina, son producto de una introducción en nuestro país.
Palabras clave: rotavirus A, Argentina, diarrea, G8P[8], G9P[4].
Abstract: Introduction. Rotavirus A (RVA) presents common and unusual G-/P- associations. The re-assortment between strains during a co-infection is one of the most important mechanisms in generating genetic diversity. In Argentina, two unusual associations (G9P[4] and G8P[8]) were detected for the first time during 2017-2018. It is unknown whether these strains represent new introductions or are the result of re-assortment between local common associations. Objective. To analyze the phylogenetic and evolutionary relationships of the G9P[4] and G8P[8] strains respect to the common associations that circulated during the same period. Materials and Methods. Representative RVA strains of both common (n=11) and unusual associations (n=2) detected during 2016-2018 were included in this study, as part of national epidemiological surveillance. Nucleic acids were extracted from fecal samples, and all 11 genomic segments were amplified and sequenced. The genetic constellations of all strains were determined, phylogenetic trees were constructed, and nucleotide and amino acid distance matrices were generated. Results. The constellations of both associations were G9P[4]-I2-R2-C2-M2-A2-N2-T2-E6-H2 and G8P[8]-I2-R2-C2-M2-A2-N2-T2-E2-H2. In 10 segments, the G8P[8] strain was more closely related to a G3P[8] strain that circulated in 2016. In 6 genes, G9P[4] was more closely related to a G2P[4] strain that circulated in 2016, whereas in 2 genes it was more closely related to a G3P[8] strain from 2016, and in 1 gene to a G2P[4] strain from 2017. However, no re-assortment events were observed in any of the 11 segments. Conclusions. The emerging G9P[4] and G8P[8] strains detected in Argentina are the result of novel introductions into our country.
Keywords: rotavirus A, Argentina, diarrhea, G8P[8], G9P[4].
Introducción
La infección por rotavirus del grupo A (RVA) es la causa más importante de diarrea aguda no inflamatoria en los niños. En general, es responsable de provocar anualmente más de 450.000 muertes en menores de 5 años, con un patrón de mortalidad heterogéneamente distribuido, ya que la mayoría de las muertes ocurren en países en desarrollo1,2. En Argentina, antes de la incorporación de la vacunación universal contra rotavirus, se notificaban anualmente más de 1,2 millones de casos de diarrea aguda de los cuales la mitad se producía en menores de 5 años3. Por su parte, el rotavirus era responsable de aproximadamente 150.000 casos de diarrea aguda, 15.000 ingresos hospitalarios y alrededor de 30 muertes por año en este grupo etario4. Por lo tanto, la enfermedad diarreica por RVA tiene un impacto significativo en la morbilidad y mortalidad, así como en los costos de salud.
El genoma del rotavirus comprende 11 segmentos de ARN bicatenario (ARNdc) que están rodeados por una cápside icosaédrica de tres capas. Estos segmentos de ARN codifican seis proteínas estructurales virales (VP1 a VP4, VP6 y VP7) y seis proteínas no estructurales (NSP1 a NSP6). La capa externa de la cápside viral contiene las proteínas VP7 y VP4, responsables de generar la producción de anticuerpos neutralizantes e, históricamente, las diferencias genéticas y antigénicas se han utilizado para clasificar a los rotavirus en tipos G (glicoproteína) y P (proteasa), respectivamente5,6.
A pesar de que se han descrito múltiples asociaciones de genotipos, solo seis han sido detectadas en forma predominante en infecciones clínicas humanas, consideradas usuales o comunes: G1P[8], G2P[4], G3P[8], G4P[8], G9P[8] y G12P[8] 7,8. Estas asociaciones de los tipos G y P han demostrado tener una mayor adaptabilidad que otras, lo que les permite diseminarse en la población de manera eficiente. La cocirculación, las fluctuaciones interanuales y geográficas, así como la aparición periódica de cepas consideradas inusuales, son características de la epidemiología del rotavirus9.
Estudios de similitud del ARN genómico de diferentes grupos y combinaciones de los tipos G y P han demostrado que existen restricciones en el reordenamiento de genes y que, en su mayoría, están reguladas por la interacción entre diferentes proteínas virales y las interacciones virus – hospedador10,11. Globalmente, se han definido tres grupos característicos (genogrupos), antigénicamente distantes entre sí, que han sido denominados de acuerdo con su cepa de referencia: Wa, DS-1 y AU-1. El genogrupo Wa presenta en su mayoría el genotipo P[8], asociado a los tipos G: G1, G3, G4, G9 y G12; el genogrupo DS-1 generalmente contiene el genotipo P[4] asociado al tipo G2; y el genogrupo AU-1 está representado por la combinación G3P[9].
Hace algo más de una década, el Grupo de Trabajo de Clasificación de Rotavirus (RCWG, del inglés, Rotavirus Classification Working Group) propuso un abordaje superador al de la clasificación binaria convencional mediante el análisis integral de los 11 segmentos del genoma de rotavirus, asignándoles a cada uno de ellos un genotipo de acuerdo con su secuencia nucleotídica, estableciendo lo que se denominan constelaciones genéticas (expresadas como GxP[x]-Ix-Rx-Mx-Cx-Ax-Nx-Tx-Ex-Hx, en donde “x” reemplaza a un número arábigo que se asigna de acuerdo con el genotipo de los genes VP7, VP4, VP6 ,VP1, VP2, VP3, NSP1, NSP2, NSP3, NSP4 y NSP5, respectivamente)12,13. Este tipo de abordaje demostró ser una herramienta útil para evaluar las relaciones evolutivas entre diferentes cepas, rastrear eventos de reordenamiento genético, estudiar la vinculación funcional de los genes y comprender los orígenes de las cepas de rotavirus poco frecuentes. En este sistema, la cepa Wa se clasifica como genotipo 1 (G1-P[8]-I1-R1-C1-M1-A1-N1-T1-E1-H1), la cepa DS-1, como genotipo 2 (G2-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2) y la cepa AU-1, como genotipo 3 (G3-P[9]-I3-R3-C3-M3-A3-N3-T3-E3-H3).
Estos virus evolucionan a través de diversos mecanismos genéticos. Debido a la falta de capacidad para corregir errores de la ARN polimerasa dependiente del ARN, los rotavirus acumulan constantemente mutaciones puntuales en cada ciclo de replicación, lo que colabora en la evolución del virus10. Por otra parte, debido a su naturaleza segmentada, el reordenamiento entre cepas de rotavirus (intercambio de uno o más segmentos genómicos entre dos cepas) como resultado de una coinfección en un mismo hospedador representa otro de los mecanismos evolutivos de mayor importancia11,14.
En el 2006, la Organización Mundial de la Salud recomendó como estrategia la incorporación de la vacunación universal contra el rotavirus en los Calendarios Nacionales de Vacunación con el objetivo de disminuir la carga de enfermedad severa asociada a este enteropatógeno viral en los menores de 5 años15. Actualmente, existen dos vacunas orales licenciadas: una pentavalente con reordenantes humano - bovino de tres dosis (RotaTeq® Merck) y una monovalente de rotavirus humano atenuado, de 2 dosis (Rotarix® GSK)16,17. Ambas vacunas han demostrado ser seguras y eficaces17,18. A pesar de que América Latina experimentó una rápida y exitosa introducción de vacunas contra el rotavirus a partir del 2006, Argentina incorporó Rotarix® GSK a su Programa Nacional de Inmunización desde el 1 de enero de 201519.
Se ha descrito que la introducción de la vacuna puede producir cambios rápidos en los patrones de circulación de los genotipos de rotavirus, es decir, cambios en la ecología viral. Particularmente, en países que habían implementado la vacuna monovalente, se observó un aumento en la detección de la asociación G2P[4] en los casos residuales de diarrea por rotavirus, y un aumento de G3P[8] cuando la vacuna pentavalente se utilizó como estrategia masiva20,21,22,23.
En Argentina, luego de dos años de la implementación de la vacuna, se observó una disminución significativa en los casos de diarrea aguda de cualquier etiología y en los casos de rotavirus confirmados en los menores de 5 años. Con respecto a los genotipos circulantes en el período posvacunal, durante el primer año , se vio un desplazamiento significativo hacia la asociación G2P[4], pero, posteriormente, disminuyó hasta hacerse indetectable y se observaron un aumento de G3P[8], G12P[8] y algunas asociaciones inusuales24. Más recientemente, se detectó un aumento en la circulación de asociaciones consideradas no comunes: G9P[4] en el 2017 y G8P[8] en el 201825. En el contexto de la vigilancia posvacunal, la emergencia de nuevas asociaciones y los nuevos paradigmas de análisis propuestos mundialmente para determinar el origen evolutivo de las mismas son hechos que promueven el abordaje de estos eventos mediante tales modelos.
El objetivo del presente trabajo fue analizar el genoma completo de las cepas inusuales emergentes de rotavirus G9P[4] y G8P[8], detectadas durante la vigilancia posvacunal en Argentina durante 2017-2018, y las relaciones filogenéticas con respecto a las cepas comunes circulantes en el mismo período. De esta forma, será posible investigar en forma integral si la emergencia de estas asociaciones inusuales son producto de un reordenamiento entre segmentos de cepas comunes que circulan frecuentemente, lo que les ha permitido adquirir una adaptabilidad eficiente para diseminarse con relativa frecuencia, o si son el resultado de la introducción de una nueva cepa.
Materiales y métodos
Se realizó un estudio descriptivo, de corte transversal, en el que se seleccionó un grupo de 13 muestras de materia fecal provenientes de la colección del Laboratorio de Gastroenteritis Virales del INEI-ANLIS “Dr. Carlos G. Malbrán” que representaran el conjunto de cepas inusuales, una G9P[4] y una G8P[8] y las comunes que circularon durante el periodo 2016 a 2018, tomando 4 muestras de cada uno de los años 2016 y 2017, y 3 muestras de 2018, que pudieron haber influenciado la adaptabilidad de ellas. Estas muestras fueron recolectadas en el marco de la vigilancia epidemiológica molecular de RVA a través de la Red Nacional de Vigilancia de Gastroenteritis Virales que coordina el Laboratorio Nacional de Referencia de Rotavirus y Norovirus. La caracterización molecular de los - tipos G y P se había realizado previamente mediante la extracción del ARN viral, a través del uso de columnas de sílica (QIAmp Viral RNA Kit, Qiagen) de acuerdo con las recomendaciones del fabricante. Posteriormente, se amplificaron y secuenciaron los genes que codifican para las proteínas VP7 y VP4, de acuerdo con procedimientos descritos previamente25. El criterio de representatividad que se utilizó en la selección fue que, para cada año, una misma combinación de tipo G/P tuviese más del 95% de identidad en el nivel nucleotídico en las secuencias de los genes VP7 y VP4 (análisis preliminar). De presentar un menor porcentaje, se incluiría para ese mismo año una segunda muestra que representara a esa subpoblación de la combinación.
Por otra parte, las muestras seleccionadas en este estudio para la genotipificación de los 9 segmentos restantes fueron reextraídas de acuerdo con lo mencionado anteriormente. Posteriormente, se realizó una retrotranscripción, seguida de una reacción en cadena de la polimerasa (RT-PCR), con el objetivo de amplificar los 9 segmentos genómicos mediante el uso de reactivos comerciales del tipo “en un solo paso” (one-step RT-PCR kit) con cebadores específicos, descritos previamente en la literatura26. Los productos de amplificación se sometieron a electroforesis en un gel de agarosa al 1% con el objetivo de visualizar las bandas correspondientes a los fragmentos esperados. Las bandas fueron cortadas del gel para luego ser purificadas por un método de extracción de gel de un kit comercial, de manera de eliminar impurezas y otros componentes de la reacción no deseados (AccuPrep PCR/Gel purification kit, Bioneer). Por último, se realizó la reacción de secuenciación utilizando los mismos cebadores específicos que se usaron en la RT-PCR. Los productos de esta reacción también fueron purificados por un kit comercial (BigDye XTerminator Purification Kit, Applied Biosystems) y se secuenciaron con un equipo de secuenciación por Sanger (3500 Series Genetic Analyzer, Applied Biosystems).
La edición de las secuencias obtenidas se realizó mediante el programa BioEdit v7.0.1. A cada una de ellas, se les asignó su genotipo correspondiente mediante el uso de la herramienta online Rotavirus A Genotype Determination, que se encuentra disponible en el sitio web Virus Pathogen Database and Analysis Resource (ViPR, https://legacy.viprbrc.org/brc/rvaGenotyper.spg?method=ShowCleanInputPage&decorator=reo). Para cada gen, se determinó el porcentaje de identidad en el nivel nucleotídico y aminoacídico de las muestras seleccionadas en el presente estudio. Asimismo, se dedujeron las secuencias de aminoácidos a partir de las secuencias nucleotídicas para identificar potenciales cambios. En ambos casos, se utilizó el programa MEGA X v10.1.8. Para conocer las relaciones evolutivas, se construyeron los árboles filogenéticos de cada gen por el método de máxima verosimilitud, mediante el servidor web IQ-TREE (http://iqtree.cibiv.univie.ac.at), posterior a la estimación del modelo de sustitución, de acuerdo con el criterio de información de Akaike y con medidas de confiabilidad del agrupamiento filogenético (Ultrafast Bootstrap).
El presente trabajo fue aprobado por el Comité de Ética en Investigación del Instituto Nacional de Enfermedades Virales Humanas INEVH-ANLIS “Dr. Julio Maiztegui”, bajo el protocolo INEVH 07/2019.
Resultados
Descripción general de la población bajo estudio
De acuerdo con los criterios de inclusión, se seleccionaron dos muestras que representaron las asociaciones inusuales (Arg15957 y Arg16571 para G9P[4] y G8P[8], respectivamente) y 11 muestras de combinaciones G-/P- comunes (G2P[4], G3P[8], G9P[8] y G12P[8]), que circularon entre los años 2016 y 2019 (Tabla 1). No se observaron subpoblaciones virales de una misma asociación para un determinado año.
Constelaciones genómicas
Las constelaciones genómicas de las cepas emergentes fueron G9P[4]-I2-R2-C2-M2-A2-N2-T2-E6-H2 y G8P[8]-I2-R2-C2-M2-A2-N2-T2-E2-H2. Por otra parte, en todas las asociaciones comunes, se observaron constelaciones puras: las cepas G2P[4] presentaron una constelación I2-R2-C2-M2-A2-N2-T2-E2-H2 y las cepas G3P[8], G9P[8] y G12P[8] poseían una disposición característica del genotipo 1 (I1-R1-C1-M1-A1-N1-T1-E1-H1). Llamativamente, 1 sola muestra (Arg15240), detectada en el año 2016, presentó la combinación G3P[8], pero con una constelación genómica pura del genotipo 2 (Tabla 1).
| Genotipo asignado | |||||||||||
| Año | Tipo G/P- | Nombre | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 |
| Cepas emergentes | |||||||||||
| 2017 | G9P[4] | Arg15957 | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E6 | H2 |
| 2018 | G8P[8] | Arg16571 | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| Asociaciones de los tipos G/P comunes | |||||||||||
| 2016 | G2P[4] | Arg15170 | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| 2016 | G3P[8] | Arg15240 | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| 2016 | G9P[8] | Arg15189 | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| 2016 | G12P[8] | Arg15133 | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| 2017 | G2P[4] | Arg15805 | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| 2017 | G3P[8] | Arg15509 | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| 2017 | G12P[8] | Arg15830 | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| 2017 | G9P[8] | Arg15892 | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| 2018 | G12P[8] | Arg16273 | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| 2018 | G9P[8] | Arg15987 | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| 2018 | G3P[8] | Arg16618 | I1 | R1 | C1 | x | A1 | N1 | T1 | E1 | H1 |
Análisis comparativo entre las cepas emergentes y las asociaciones comunes
En el análisis del gen VP7, se detectaron 5 tipos G: G2, G3, G8, G9 y G12. De acuerdo con lo esperado por representar un genotipo inusual, G8 demostró una identidad promedio en los niveles nucleotídico y aminoacídico de 74,1% y 81,0%, respectivamente y no se agrupó con ninguno de los genotipos considerados comunes en el árbol filogenético (Figura 1). Por otra parte, el genotipo G9 de la asociación G9P[4] se agrupó con la cepa Arg15189 (G9P[8]) que circuló en el año 2016. Asimismo, entre ellas se observó la mayor similitud, con una identidad de 96,6% en el nivel nucleotídico y también aminoacídico (Figura 1). Con respecto al análisis comparativo de la secuencia de aminoácidos, se identificaron diez cambios aminoacídicos, de los cuales solo uno corresponde a un sitio antigénico (E125K).
Por otra parte, en el análisis de VP4 se detectaron 2 tipos P: P[4] y P[8]. Se observó que la P[4] detectada en el 2017 se agrupa con 2 cepas asociadas con el genotipo G2: una que circuló ese mismo año (Arg15805), y otra detectada el año anterior (Arg15170). Sin embargo, con la que presentó mayor similitud en el nivel nucleotídico y el aminoacídico fue con Arg15170, con identidades nucleotídicas de 97,4% y aminoacídicas de 97,3%, respectivamente. El análisis de las secuencias de aminoácidos reveló siete diferencias, de las cuales solo una corresponde a un sitio antigénico (Q114P). Por otro lado, la P[8] se agrupó con una G3P[8], Arg15240, que circuló en nuestro país en el 2016, con una similitud nucleotídica y aminoacídica de 98,9% y 97,9%, respectivamente (Figura 2). En este caso, solo se identificaron 3 cambios de aminoácidos entre ambas secuencias, de los cuales uno se relaciona con un sitio antigénico (G195D)
Con respecto al resto de los 9 segmentos genómicos, se observaron algunas similitudes en el patrón de agrupamientos. En los árboles filogenéticos de los genes VP6, VP3, NSP3 y NSP5, la cepa G8P[8] se agrupó en un clado separado del resto de las cepas que también poseían el genotipo I2, M2, T2 y H2, respectivamente (Figura 3, Figura 6, Figura 9, y Figura 11). Desafortunadamente, no se pudo asignar el tipo M de la cepa Arg16618 debido a que, luego de múltiples intentos, no fue posible amplificar el gen VP3. De cualquier manera, la cepa emergente presentó mayor identidad en el nivel nucleotídico y de aminoácidos para estos cuatro genes con la cepa G3P[8] Arg15240 que circuló en el año 2016. Por el contrario, en la cepa G9P[4], se observó una relación evolutiva más cercana a las cepas G2P[4] en estos genes. En términos de similitud, en los genes NSP3 y NSP5 se observó la mayor identidad con la cepa G2P[4] Arg15170 que circuló en el año 2016. Sin embargo, llamativamente, en los genes VP6 y VP3, la cepa G9P[4] mostró mayor similitud nucleotídica y aminoacídica con la otra asociación emergente, G8P[8] (Figura 3, Figura 6, Figura 9, y Figura 11).
El análisis del gen VP1 mostró un patrón de agrupamiento distinto. Si bien G8P[8] se agrupó en un clado separado de las otras cepas con genotipo R2, se observó que G9P[4] se agrupó junto con las cepas G2P[4] y G3P[8] (Figura 4). Esto también se observó analizando el gen NSP2 (Figura 8). Las mayores similitudes en VP1 se encontraron con la cepa Arg15805 (G2P[4]) y la cepa Arg15240 (G3P[8]) para G9P[4] y G8P[8], respectivamente (Figura 4). Con respecto a NSP2, tanto la cepa G8P[8] como la cepa G9P[4] presentaron el mayor porcentaje de similitud con la cepa G3P[8] Arg15240 (Figura 8).
Los árboles filogenéticos de los genes VP2 y NSP1 demostraron que tanto G8P[8] como G9P[4] se agruparon en el mismo subclado del genotipo C2 y A2, respectivamente, junto con la cepa Arg15240 (G3P[8] del año 2016) (Figuras 5 y 7). Sin embargo, el análisis de identidad demostró que solo la cepa G8P[8] presentó el mayor porcentaje de similitud con la cepa Arg15240, mientras que G9P[4] tuvo mayor similitud con las cepas G2P[4] (Figura 5 y Figura 7).
Por último, el estudio del gen NSP4 demostró la presencia de 3 tipos E: E1, E2 y E6. Nuevamente, el genotipo de la G9P[4] (E6) no se agrupó con ninguna de las secuencias de las cepas comunes por representar un genotipo distinto. En cambio, se observó que la cepa G8P[8] estaba más cercanamente relacionada con la cepa Arg15240 (G3P[8]) tanto en términos evolutivos como en la identidad nucleotídica y aminoacídica (Figura 10).
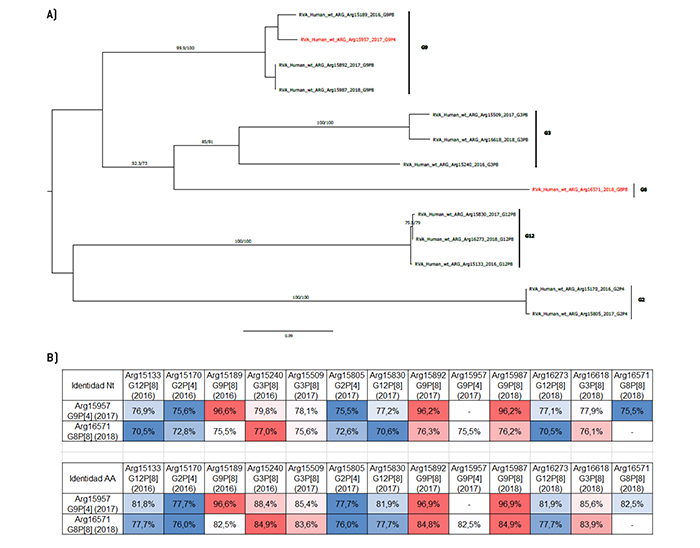
Figura 1.
Análisis filogenético y matriz de distancias del gen VP7
a) Árbol filogenético de máxima verosimilitud del gen VP7 del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos G se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen VP7 de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
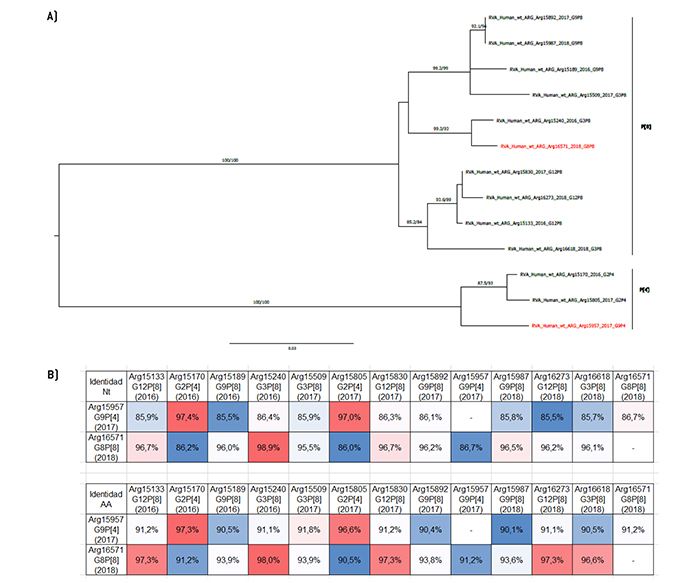
Figura 2.
Análisis filogenético y matriz de distancias del gen VP4.
a) Árbol filogenético de máxima verosimilitud del gen VP4 (parcial) del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de aso-ciaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos P se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen VP4 (parcial) de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
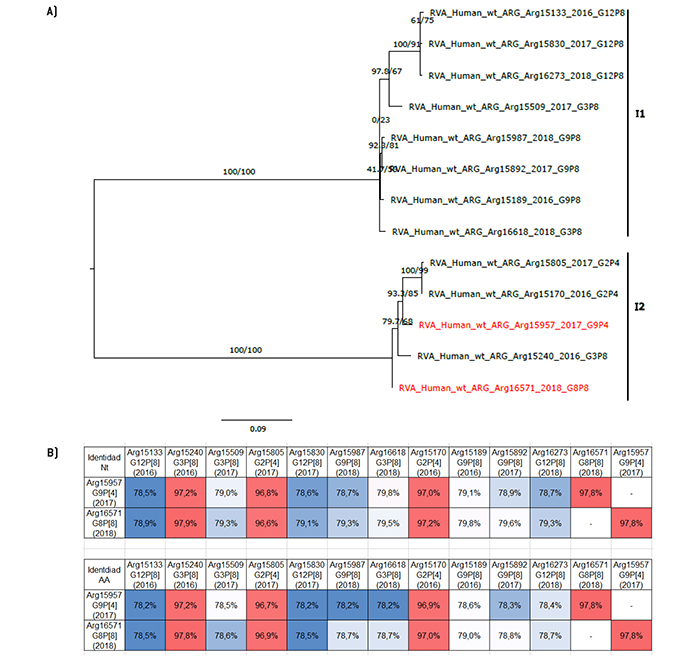
Figura 3.
Análisis filogenético y matriz de distancias del gen VP6
a) Árbol filogenético de máxima verosimilitud del gen VP6 del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos I se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen VP6 de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
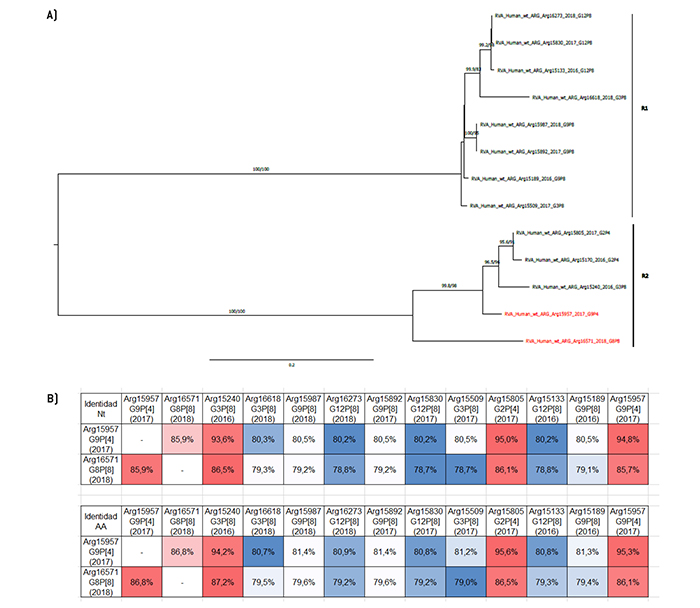
Figura 4.
Análisis filogenético y matriz de distancias del gen VP1
a) Árbol filogenético de máxima verosimilitud del gen VP1 (parcial) del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos R se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen VP1 (parcial) de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
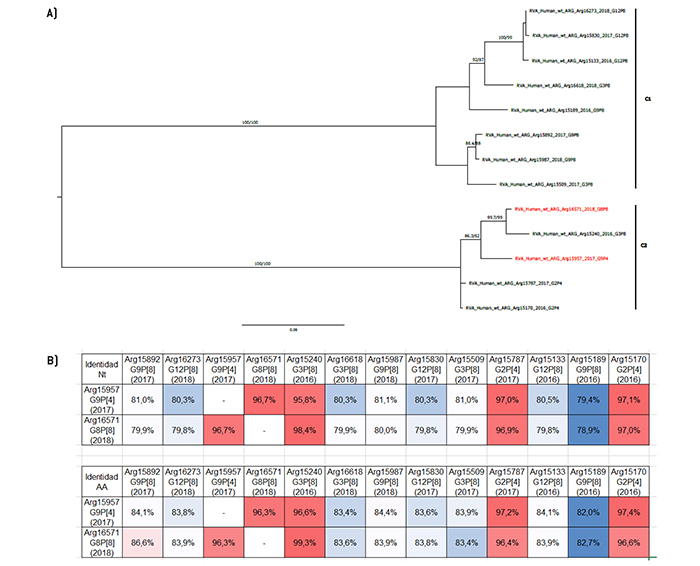
Figura 5.
Análisis filogenético y matriz de distancias del gen VP2
a) Árbol filogenético de máxima verosimilitud del gen VP2 (parcial) del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos C se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen VP2 (parcial) de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
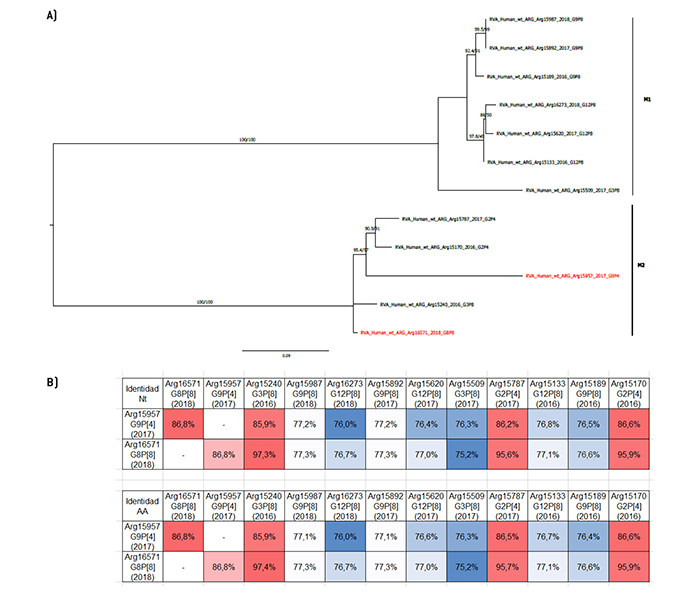
Figura 6.
Análisis filogenético y matriz de distancias del gen VP
a) Árbol filogenético de máxima verosimilitud del gen VP3 (parcial) del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos M se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen VP3 (parcial) de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
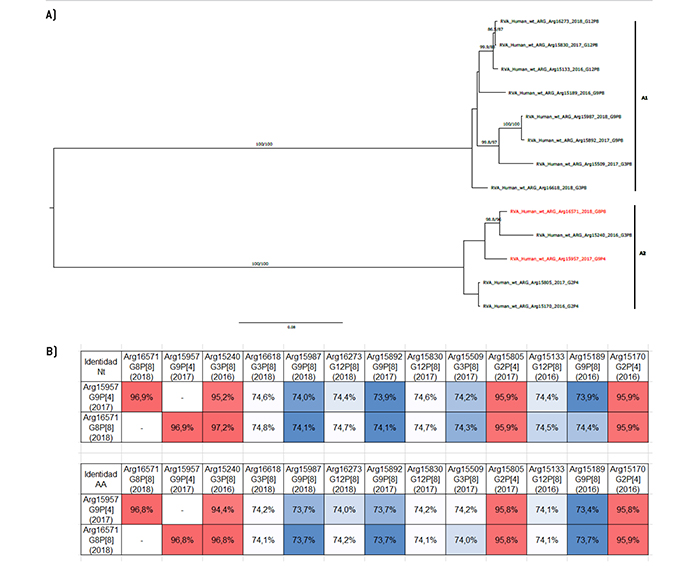
Figura 7.
Análisis filogenético y matriz de distancias del gen NSP1
a) Árbol filogenético de máxima verosimilitud del gen NSP1 del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos A se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen NSP1 de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
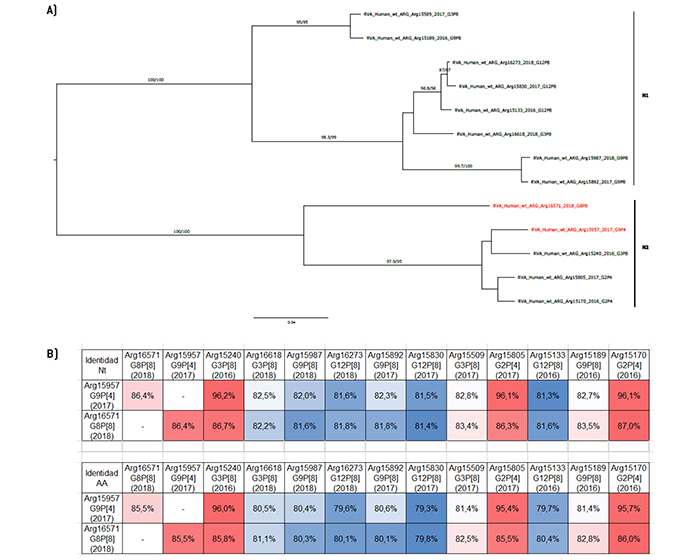
Figura 8.
Análisis filogenético y matriz de distancias del gen NSP2
a) Árbol filogenético de máxima verosimilitud del gen NSP2 del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos N se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen NSP2 de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
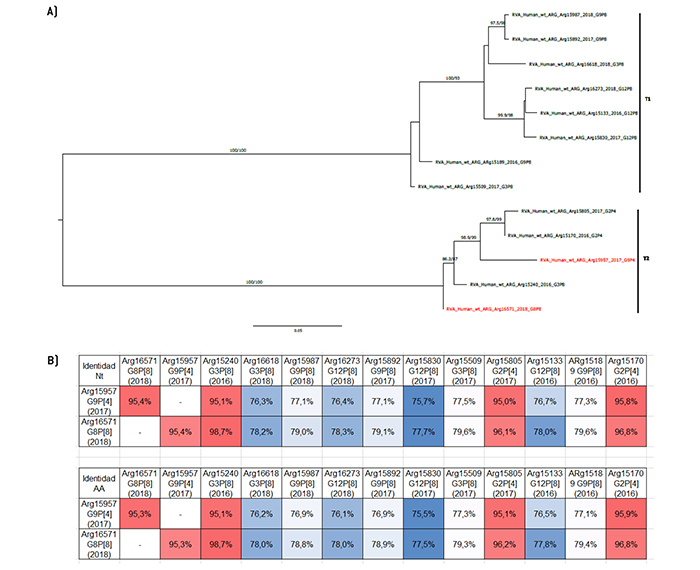
Figura 9.
Análisis filogenético y matriz de distancias del gen NSP3
a) Árbol filogenético de máxima verosimilitud del gen NSP3 del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos T se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen NSP3 de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
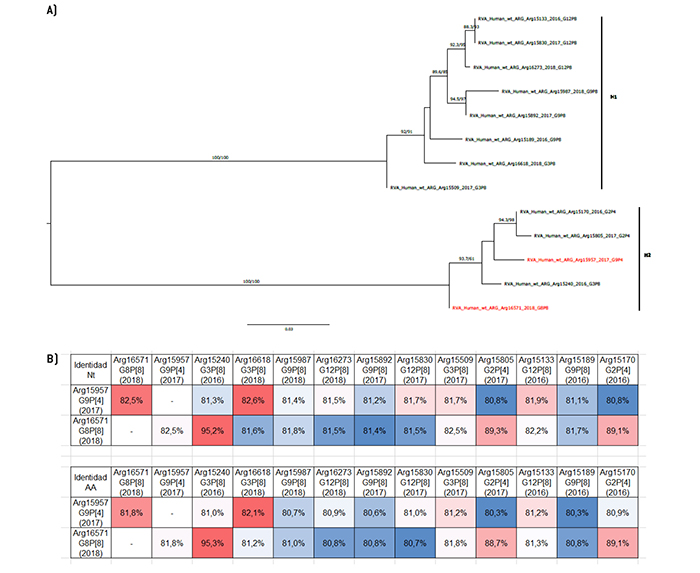
Figura 10.
Análisis filogenético y matriz de distancias del gen NSP4
a) Árbol filogenético de máxima verosimilitud del gen NSP4 del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos E se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen NSP4 de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes.
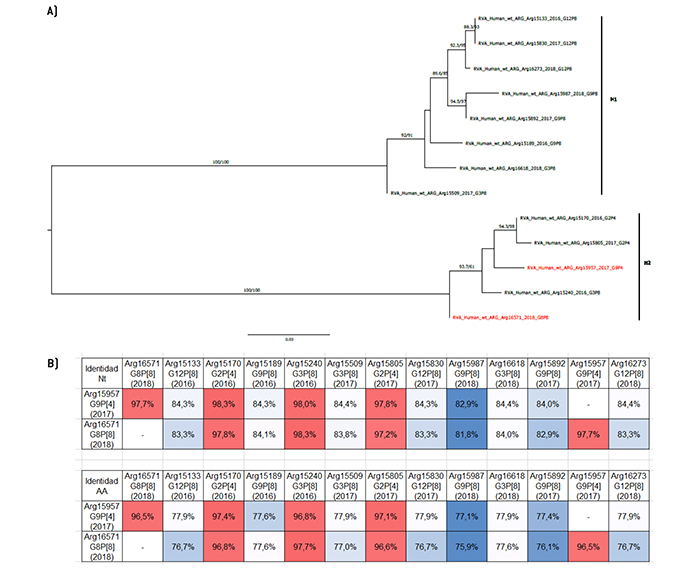
Figura 11.
Análisis filogenético y matriz de distancias del gen NSP5
a) Árbol filogenético de máxima verosimilitud del gen NSP5 del rotavirus de las cepas emergentes G9P[4] y G8P[8], y de cepas de asociaciones comunes que circularon durante el período 2016-2018 en Argentina. Se muestran los valores de Ultrafast Bootstrap (>85) a partir de 10.000 réplicas en los nodos. Las barras de escala indican la distancia genética (sustituciones de nucleótidos por sitio). Las cepas emergentes se indican en color rojo y los distintos tipos H se muestran en llaves. b) Tabla de porcentaje de identidad en los niveles nucleotídico (nt) y aminoacídico (aa) del gen NSP5 de las cepas emergentes G9P[4] y G8P[8] con respecto a las cepas de asociaciones comunes
Discusión
El presente estudio analizó la constelación genética de las cepas inusuales G9P[4] y G8P[8], junto con otras asociaciones comunes que circularon contemporáneamente. Este abordaje resulta de utilidad para conocer si existieron potenciales eventos de reordenamiento que les hayan permitido a estas cepas emergentes adquirir adaptabilidad y expliquen su circulación en moderada frecuencia12,27
G8P[8] se relacionó mayoritariamente con una cepa G3P[8]-equine like que circuló en el año 2016. Por otra parte, G9P[4] demostró tener mayor heterogeneidad en su identidad, aunque principalmente se observó que está más evolutivamente relacionada con cepas G2P[4]. De cualquier manera, en ninguno de los once segmentos se alcanzó una similitud por encima del 99%. Estos resultados indican que estas asociaciones no adquirieron segmentos de las cepas comunes locales. Por lo tanto, la emergencia de G9P[4] y G8P[8] fue producto de una nueva introducción. En este sentido, estudios previos realizados solamente con el gen VP7 revelaron que ambas combinaciones inusuales mostraron mayor identidad con cepas que fueron identificadas circulando en Europa y el sudeste asiático25. Estos resultados extienden esta observación a los otros diez segmentos genómicos restantes.
El análisis integral de las constelaciones genéticas ha permitido determinar que hay combinaciones de proteínas que funcionan mejor que otras y que le brindan al virus mayor adaptabilidad y eficiencia en su diseminación10,27. Este fenómeno, denominado vinculación genética, se observa en las cepas emergentes detectadas. En primer lugar, no se observaron constelaciones mixtas entre los genotipos 1 y 2. G9P[4] es una combinación de un tipo G y un P , a los que habitualmente se los asocia con orígenes evolutivos distintos (Wa-like y DS-1-like, respectivamente). Sin embargo, la presencia de una constelación asociada al genotipo 2 indicaría que la vinculación genética más favorable estuvo determinada por el tipo P. Por otra parte, G8P[8] retrata un escenario inverso: un tipo Gasociado a un origen DS-1-like y un tipo Pa Wa-like. Pero, en este caso, este tipo Pya ha demostrado asociarse con constelaciones genéticas puras del genotipo 2 de forma eficiente28,29, por lo que podría extenderse la hipótesis de una configuración genética determinada por el gen VP4 (tipo P).
Si bien la estrategia de vacunación universal resultó ser exitosa en términos de reducir la carga de enfermedad diarreica en los menores de 5 años, la detección de estos nuevos emergentes, genéticamente distantes de la cepa vacunal, en los casos residuales, sugiere el papel que las vacunas cumplen en la presión selectiva. Asimismo, la creciente frecuencia con la que se identifican las asociaciones inusuales DS-1-like demuestran que pudieron haber desarrollado mecanismos más eficientes de diseminación, en un contexto de disminución de poblaciones susceptibles a cepas Wa-like. Por lo tanto, este análisis representa un punto de partida para futuros estudios de adaptabilidad de este tipo de emergentes.
El presente trabajo contempla algunas limitaciones. En primer lugar, si bien se exploró la diversidad de cada asociación para el período de estudio, no se descarta que circulen distintas variantes de la misma asociación tipo G/Pen una misma temporada. Del mismo modo, en este estudio, se contemplaron las cepas circulantes detectadas entre 2016 y 2018. Por lo tanto, podría esperarse que algunos de los segmentos de la constelación de las cepas inusuales pudieran haberse adquirido de otras que circularon con anterioridad y persistieron en la población de forma críptica.
De cualquier manera, los resultados obtenidos en este estudio son de gran utilidad, ya que aportan evidencia de la compleja dinámica de la circulación de los genotipos de RVA en nuestro país. Además, contribuyen con información sustancial para los responsables implicados en la toma de decisiones respecto del impacto de la vacuna contra nuevos emergentes. Se considera necesario analizar en profundidad estos eventos evolutivos en el marco de una vigilancia local y continua de la diversidad genética luego de esta intervención para analizar su impacto en la población pediátrica no solo desde el punto de vista clínico, sino también de la ecología viral.
Agradecimientos
Los autores agradecen a los integrantes de la Red Nacional de Vigilancia por Laboratorios de Gastroenteritis Virales por su participación en la recolección de muestras clínicas y su compromiso con la vigilancia epidemiológica de rotavirus.
Referencias bibliográficas
1. Organización Mundial de la Salud. Centro de prensa. Enfermedades dia-rreicas. 2009; nota descriptiva n º 330. [Acceso: 4 de junio de 2012]. Disponible en: http://www.who.int/mediacentre/factsheets/fs330/es/
2. Tate JE, Burton AH, Bosch I, Pinto C, Steele AD, Duque J, et al. 2008 estimate of worldwide rotavirus-associated mortality in children youn-ger than 5 years before the introduction of universal rotavirus vacci-nation programmes: a systematic review and meta-analysis. Lancet Infect Dis 2012;12(2):136-141, https://doi.org/10.1016/s1473- 3099(11)70253-5
3. Ministerio de Salud de la Nación. Boletín Epidemiológico Anual 2010. Argentina, 2011. [Acceso: 4 de junio de 2012]. Disponible en http:// msal.gov.ar/htm/site/sala_situacion/PANELES/bep-anual-2010/BE-PANUAL_2010.pdf
4. Degiuseppe JI, Giovacchini C, Stupka JA; Red Nacional de Vigilancia de Gastroenteritis Virales. 2013. Rotavirus epidemiology and surveillan-ce in Argentina: 2009 - 2011 Arch Arg Pediatr 2013;111(2):148-54, http://dx.doi.org/10.5546/aap.2013.148
5. Greenberg HB, Estes MK. Rotaviruses: from pathogenesis to vac-cination. Gastroenterology. 2009; 136(6):1939-51, https://doi. org/10.1053/j.gastro.2009.02.076
6. Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Bányai K, Brister JR, et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch Virol. 2011; 156(8):1397-413, https://doi.org/10.1007/s00705-011-1006-z
7. Trojnar E, Sachsenröder J, Twardziok S, Reetz J, Otto PH, Johne R. Iden-tification of an avian group A rotavirus containing a novel VP4 gene with a close relationship to those of mammalian rotaviruses. J Gen Vi-rol. 2013; 94(Pt 1):136-142, https://doi.org/10.1099/vir.0.047381-0
8. Degiuseppe JI, Beltramino JC, Millán A, Stupka JA, Parra GI. Complete genome analyses of G4P[6] rotavirus detected in Argentinean chil-dren with diarrhoea provides evidence of interspecies transmission from swine. Clin Microbiol Infect. 2013; 19(8):E367-71, https://doi. org/10.1111/1469-0691.12216
9. Martella V, Banyai K, Matthijnssens J, Buonavoglia C, Ciarlet M. 2009. Zoonotic aspects of rotaviruses. Vet Microbiol. 2010;140(3-4):246- 255, https://doi.org/10.1016/j.vetmic.2009.08.028
10. McDonald SM, Matthijnssens J, McAllen JK, Hine E, Overton L, Wang S, et al. Evolutionary dynamics of human rotaviruses: balancing reas-sortment with preferred genome constellations. PLoS Pathog. 2009; 5(10):e1000634, https://doi.org/10.1371/journal.ppat.1000634
11. Desselberger U. Genome rearrangements of rotaviruses. Arch Virol Suppl. 1996;12:37-51, https://doi.org/10.1007/978-3-7091-6553-9_5
12. Matthijnssens J, Ciarlet M, Heiman E, Arijs I, Delbeke T, McDonald SM, et al. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and hu-man DS-1-like and bovine rotavirus strains. J Virol. 2008; 82(7):3204- 19, https://doi.org/10.1128/jvi.02257-07
13. Matthijnssens J, Van Ranst M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr Opin Virol. 2012; 2(4):426- 33, https://doi.org/10.1016/j.coviro.2012.04.007
14. Matthijnssens J, Bilcke J, Ciarlet M, Martella V, Bányai K, Rahman M, et al. Rotavirus disease and vaccination: impact on genotype diversity. Future Microbiol. 2009; 4(10):1303-16, https://doi.org/10.2217/fmb.09.96
15. World Health Organization. Rotavirus vaccines: an update. Weekly Epi-demiological Record 84, 533 – 540 2009.
16. Ruiz-Palacios GM, Pérez-Schael I, Velázquez FR, Abate H, Breuer T, Clemens SC, et al; Human Rotavirus Vaccine Study Group. Safety and efficacy of an attenuated vaccine against severe rotavirus gastroen-teritis. N Engl J Med. 2006;354(1):11-22, https://doi.org/10.1056/ nejmoa052434
17. Vesikari T, Matson DO, Dennehy P, Van Damme P, Santosham M, Rodri-guez Z, et al; Rotavirus Efficacy and Safety Trial (REST) Study Team. Safety and efficacy of a pentavalent human-bovine (WC3) reassortant rotavirus vaccine. N Engl J Med. 2006 Jan 5;354(1):23-33, https://doi. org/10.1056/nejmoa052664
18. Velázquez RF, Linhares AC, Muñoz S, Seron P, Lorca P, DeAntonio R, et al. Efficacy, safety and effectiveness of licensed rotavirus vaccines: a systematic review and meta-analysis for Latin America and the Caribbean. BMC Pediatr. 2017;17(1):14, https://doi.org/10.1186/ s12887-016-0771-y
19. de Oliveira LH, Danovaro-Holliday MC, Sanwogou NJ, Ruiz-Matus C, Tambini G, Andrus JK. Progress in the introduction of the rotavirus vaccine in Latin America and the Caribbean: four years of accumulated experience. Pediatr Infect Dis J. 2011; 30(1 Suppl):S61-6, https://doi.org/10.1097/inf.0b013e3181fefdd6
20. Kirkwood CD, Boniface K, Barnes GL, Bishop RF. Distribution of rotavirus genotypes after introduction of rotavirus vaccines, Rotarix® and RotaTeq®, into the National Immunization Program of Australia. Pediatr Infect Dis J. 2011; 30(1 Suppl):S48-53, https://doi.org/10.1097/inf.0b013e3181fefd90
21. Hull JJ, Teel EN, Kerin TK, Freeman MM, Esona MD, Gentsch JR, et al; National Rotavirus Strain Surveillance System. United States rotavirus strain surveillance from 2005 to 2008: genotype prevalence before and after vaccine introduction. Pediatr Infect Dis J. 2011; 30(1 Suppl):S42-7, https://doi.org/10.1097/inf.0b013e3181fefd78
22. Gurgel RQ, Cuevas LE, Vieira SC, Barros VC, Fontes PB, Salustino EF, et al. Predominance of rotavirus P[4]G2 in a vaccinated population, Brazil. Emerg Infect Dis. 2007;13(10):1571-3, https://doi.org/10.3201/eid1310.070412
23. Zeller M, Rahman M, Heylen E, De Coster S, De Vos S, Arijs I, et al. Rotavirus incidence and genotype distribution before and after national rotavirus vaccine introduction in Belgium. Vaccine. 2010 Nov 3;28(47):7507-13, https://doi.org/10.1016/j.vaccine.2010.09.004
24. Degiuseppe JI, Stupka JA. First assessment of all-cause acute diarrhoea and rotavirus-confirmed cases following massive vaccination in Argentina. Epidemiol Infect. 2018; 146(15):1948-1954, https://doi.org/10.1017/s0950268818001954
25. Degiuseppe JI, Stupka JA; Argentinean Rotavirus Surveillance Network. Emergence of unusual rotavirus G9P[4] and G8P[8] strains uring post vaccination surveillance in Argentina, 2017-2018. Infect Genet Evol. 2021;93:104940, https://doi.org/10.1016/j.meegid.2021.104940
26. Fujii Y, Shimoike T, Takagi H, Murakami K, Todaka-Takai R, Park Y, et al. Amplification of all 11 RNA segments of group A rotaviruses based on reverse transcription polymerase chain reaction. Microbiol Immunol 2012;56(9):630-8, https://doi.org/10.1111/j.1348-0421.2012.00479.x
27. Heiman EM, McDonald SM, Barro M, Taraporewala ZF, Bar-Magen T, Patton JT. Group A human rotavirus genomics: evidence that gene constellations are influenced by viral protein interactions. J Virol. 2008; 82(22):11106-16, https://doi.org/10.1128/jvi.01402-08
28. Mwangi PN, Mogotsi MT, Rasebotsa SP, Seheri ML, Mphahlele MJ, NdzeVN, et al. Uncovering the First Atypical DS-1-like G1P[8] Rotavirus Strains That Circulated during Pre-Rotavirus Vaccine Introduction Era in South Africa. Pathogens. 2020; 9(5):391, https://doi.org/10.3390/pathogens9050391
29. Tacharoenmuang R, Komoto S, Guntapong R, Upachai S, Singchai P, I de T, Fukuda S, et al. High prevalence of equine-like G3P[8] rotavirus in children and adults with acute gastroenteritis in Thailand. J Med Virol. 2020;92(2): 174-186, https://doi.org/10.1002/jmv.25591
Notas de autor
antomartelli94@gmail.com
Información adicional
redalyc-journal-id: 651