Revisión
Hipergammaglobulinemia: su relación con distintos estados clínico-patológicos y el rol del laboratorio
Hypergammaglobulinemia: its relationship with different clinical-pathological states and the role of the laboratory


Hipergammaglobulinemia: su relación con distintos estados clínico-patológicos y el rol del laboratorio
Bioquímica y Patología Clínica, vol. 89, núm. 3, pp. 52-58, 2025
Asociación Bioquímica Argentina
Resumen: Introducción: La hipergammaglobulinemia es el aumento en la producción de inmunoglobulinas por linfocitos B en respuesta a distintos estímulos. Se asocia a varios estados clínico-patológicos. Objetivo: Describir la utilidad de la detección de la hipergammaglobulinemia, su relación con distintos estados clínico-patológicos y el rol del laboratorio. Materiales y métodos: Se realizó una búsqueda bibliográfica en PubMed y Google Académico. Desarrollo: Para detectar la hipergammaglobulinemia, se realizan distintas técnicas, como la electroforesis de proteínas para un análisis cualitativo, las inmunoglobulinas que migran en el sector gamma, la inmunofijación o inmunoelectroforesis para clasificarla en monoclonal o policlonal y técnicas cuantitativas para determinar concentraciones de las distintas clases. La hipergammaglobulinemia policlonal se presenta en infecciones virales crónicas, patologías autoinmunes y errores innatos de la inmunidad, mientras que la monoclonal se asocia a neoplasias malignas. Conclusión: Mediante distintas técnicas de laboratorio, se puede identificar la hipergammaglobulinemia y, gracias a esto, contribuir al diagnóstico del paciente y a la diferenciación de otros estados clínicos patológicos.
Palabras clave: hipergammaglobulinemia, proteinograma por electroforesis, inmunoglobulinas, gammapatía monoclonal, gammapatía policlonal, inmunofijación.
Abstract: Background: Hypergammaglobulinemia is the increase in the production of immunoglobulins by B Lymphocytes in response to different stimuli. It is associated with several clinical-pathological states. Objective: To describe the usefulness of the detection of hypergammaglobulinemia, its relationship with different clinical-pathological states, and the role of the laboratory. Materials and methods: A bibliographic search was carried out in Pubmed and Google Scholar. Results showed that hypergammaglobulinemia is detected through different techniques, such as protein electrophoresis for a qualitative analysis of immunoglobulins that migrate in the gamma sector, immunofixation or immunoelectrophoresis to classify it as monoclonal or polyclonal, and quantitative techniques to determine concentrations of the different classes. Results also showed that polyclonal hypergammaglobulinemia occurs in chronic viral infections, autoimmune pathologies and inborn errors of immunity, while monoclonal hypergammaglobulinemia is associated with malignant neoplasms. Conclusion: Hypergammaglobulinemia can be identified through different laboratory techniques and, thanks to this, it can contribute to the diagnosis of the patient and the differentiation with other pathological clinical states.
Keywords: hypergammaglobulinemia, proteinogram by electrophoresis, immunoglobulins, monoclonal gammopathy, polyclonal gammopathy, immunofixation.
Introducción
Las inmunoglobulinas (Igs), constituyentes del suero, se generan a partir de la activación y diferenciación de linfocitos B (LB) tras la exposición a antígenos1,2. Existen cinco clases: IgG, IgA, IgM, IgE e IgD, cada una con funciones específicas en la respuesta inmune1,2,3. La hipergammaglobulinemia (Hγg) resulta de la sobreproducción de Igs en respuesta a estímulos, ya sean infecciosos, autoinmunes o neoplásicos4,5.
El perfil densitométrico del proteinograma por electroforesis (PxE) puede revelar la presencia de Hγg y clasificarla como policlonal o monoclonal, según la morfología del pico observado en la región gamma, comigrando en beta-1, beta-2, alfa-2; o, entre las fracciones. La presencia de una zona heterogénea, ancha y difusa en la zona gamma del PxE sugiere una Hγg policlonal (Figura 1) debido a la expansión de diversos clones de LB, asociada a causas reactivas, autoinmunes o errores innatos de la inmunidad. La presencia, en cambio, de una banda homogénea, alta, densa y delgada, en las regiones alfa, beta o gamma del PxE corresponde a una Hγg monoclonal (Figura 2). Este hallazgo orienta hacia una gammapatía monoclonal (GM), caracterizada por la proliferación de un solo clon de LB, que requiere estudios adicionales como inmunofijación, cuantificación de cadenas livianas libres y punción de médula ósea (MO) para la evaluación de la clonalidad3,5,6,7.
Los objetivos de este estudio fueron describir la utilidad de la hipergammaglobulinemia para el diagnóstico de distintos estados clínico-patológicos y destacar el rol del laboratorio para identificar la hipergammaglobulinemia mediante distintas técnicas.
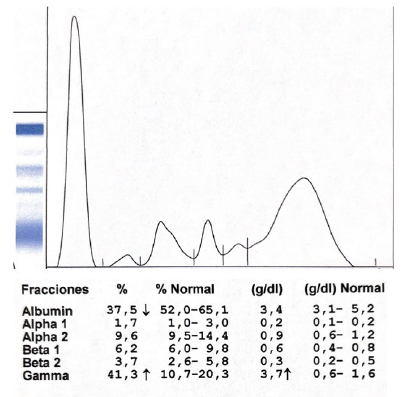
Figura 1
Pico policlonal.
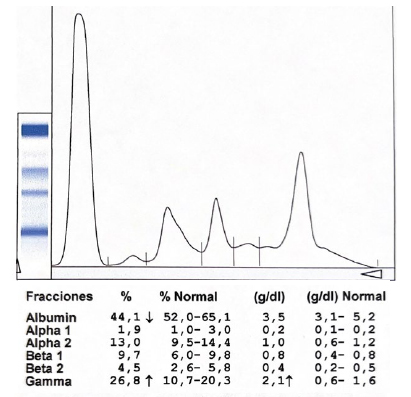
Figura 2
Pico monoclonal
Materiales y métodos
Se realizó un estudio de revisión bibliográfica a partir de la búsqueda, selección y análisis de artículos científicos. Se llevaron a cabo búsquedas en la base de datos MEDLINE a través de su buscador PubMed y en el buscador Google académico. Se utilizaron los términos: hipergammaglobulinemia, gammapatías monoclonales, proteinograma por electroforesis, maduración del sistema inmune, técnicas para determinar inmunoglobulinas, técnicas del laboratorio de inmunología, hipergammaglobulinemia en infecciones virales, hipergammaglobulinemia en neoplasias malignas.
Se seleccionaron publicaciones de los últimos diez años y publicaciones más antiguas que a las que se hace referencia habitualmente y que gozan de gran prestigio.
Se incluyeron todo tipo de artículos: trabajos de investigación, revisiones, capítulos de libros, recomendaciones y protocolos de laboratorio, etc.. Dentro de estos, se seleccionaron aquellos de revistas con mayor impacto científico, de autores reconocidos, con información actualizada y de calidad con respecto al tema investigado.
Se excluyeron artículos de bajo impacto científico, trabajos de investigación con tamaños muestrales pequeños, autores o centros con menor experiencia en el tema.
| IgG (mg/dl) | IgA (mg/dl) | IgM (mg/dl) | |
| RN (término) | 610-1.500 | 1-4 | 6-30 |
| 3 meses | 170-560 | 5-50 | 30-100 |
| 6 meses | 200-670 | 8-70 | 30-100 |
| 1 año | 330-1.160 | 10-100 | 40-170 |
| 2-6 años | 400-1.100 | 10-160 | 50-180 |
| 7-12 años | 600-1.230 | 30-200 | 50-200 |
| Adultos | 680-1.530 | 66-400 | 36-260 |
Desarrollo
Maduración del sistema inmune y producción de gammaglobulinas
La producción de inmunoglobulinas está vinculada a la maduración del sistema inmune desde el nacimiento hasta la adolescencia y se refleja en la variación de valores de los distintos isotipos de Igs. Conocer los valores de referencia de Igs según la edad es crucial para identificar la Hγg9.
Los LB productores de inmunoglobulinas provienen de las células precursoras del sistema hematopoyético, maduran en MO y se diferencian a células plasmáticas (CP) en tejidos secundarios9.
Ontogénicamente, se producen múltiples cambios en los niveles de Igs desde el nacimiento hasta los ocho o diez años, en que se estabilizan y alcanzan los valores de Igs del adulto2,9,10.
El recién nacido tiene un número elevado de LB, pero mínima diferenciación de CP. Por este motivo, el nivel de todas las Igs es bajo, excepto IgG gracias al paso transplacentario. Conforme pasan los primeros meses de vida, los niveles de IgG disminuyen en forma progresiva creando un bache inmunológico hasta los 6 meses, momento en el cual comienza a sintetizar IgG2,9,10.
La IgM, única Ig sintetizada por los neonatos normalmente, aumenta rápidamente después del primer mes de vida como respuesta a la colonización intestinal10.
La IgA, inicialmente deficiente en el epitelio mucoso intestinal fetal, aumenta en recién nacidos por el aporte del calostro y la leche materna10. Ver valores de referencia en Tabla I.
Metodologías para la detección de hipergammaglobulinemia
Las alteraciones cualitativas o cuantitativas de las inmunoglobulinas forman parte del cuadro clínico de numerosas enfermedades. En este sentido, resalta la importancia de cuantificarlas mediante diferentes metodologías7.
Electroforesis de proteínas
Esta técnica consiste en la migración de las proteínas del suero a través de un gel (o matriz porosa) en el cual, por acción de un campo eléctrico, serán separadas de acuerdo con su carga eléctrica y peso molecular. De esta manera, se obtienen 5 bandas proteicas: albúmina, alfaglobulinas de tipo 1 y 2, betaglobulinas de tipo 1 y 2 y una zona, la de las gammaglobulinas. También permite calcular la concentración de cada una respecto de la concentración de proteínas totales11,12.
La electroforesis puede realizarse en soporte sólido donde, luego de la separación, se tiñen con colorantes y se cuantifican las fracciones por medio de un densitómetro12,13,14.
Por otro lado, en la electroforesis capilar, se realiza la separación de proteínas en un capilar, donde, por la aplicación de un campo eléctrico, se produce un flujo electroendosmótico que excede la movilidad electroforética por lo que las proteínas migran. Luego de la separación, se usan distintos detectores para cuantificar las proteínas. Al comparar este método con la electroforesis de proteínas en soporte sólido, se observa que posee varias ventajas: elevada resolución, mayor reproducibilidad, precisión y sensibilidad, automatización, rapidez, menores volúmenes de muestra y bajo costo12,13.
La electroforesis de orina concentrada se usa para detectar la eliminación urinaria de cadenas livianas libres monoclonales (CLLM); estas son fragmentos de Igs que se exponen cuando no están unidas, por lo que se las denomina libres. La eliminación urinaria se denomina proteinuria de Bence Jones, y está presente en GM. Esta técnica aporta información sobre el índice de masa tumoral y permite su monitorización7.
Inmunofijación
Consiste en realizar la separación de las proteínas mediante una electroforesis de suero en carriles diferentes. Luego, se siembran antisueros específicos contra IgG, IgA, IgM, kappa y lambda que precipitan con la proteína y, después, se tiñen con un colorante específico para revelar las bandas reconocidas7,11,13,14. Esta técnica es sencilla, rápida, sensible, clara y fácilmente interpretable por lo que prácticamente sustituyó a la inmunoelectroforesis2,13.
Se utiliza principalmente para GM, ya que permite caracterizar el tipo de cadena pesada y liviana que compone la paraproteína. Sin embargo, no permite cuantificar la Hγg. Se realiza en suero y, además, en orina concentrada, ya que es más sensible y específica que otros ensayos más antiguos de Bence Jones2,7,11.
Nefelometría y turbidimetría
Esta metodología se basa en agregar a la muestra de suero un exceso del anticuerpo (Ac) específico contra las Igs que se quieren determinar. Se forman agregados que dan turbidez y luego, un precipitado que dispersa la luz13,14,15.
En la turbidimetría, el detector mide el descenso de la luz transmitida por la dispersión por las partículas, mientras que, en la nefelometría, en cambio, se mide la luz dispersada11,13.
Estas técnicas permiten cuantificar la proteína de manera rápida y automatizada. Sin embargo, como desventaja, pueden existir interferencias por turbidez de la muestra7,14,15.
Por otro lado, dichas técnicas se utilizan para medir CLLM en casos de Hγg monoclonal en suero y orina mediante Acs contra estas. Se calcula la razón K/l: un índice alterado orienta a una producción clonal de CLLM11.
Se usan para diagnóstico (Dco.) y seguimiento de pacientes con enfermedad renal con gammapatías11.
Quimioluminiscencia
Estos tipos de ensayos se utilizan para la detección de IgE debido a su alta sensibilidad, que permite su cuantificación, ya que la misma se encuentra en concentraciones muy bajas en suero15.
En los inmunoensayos quimioluminiscentes, se emplean moléculas que emiten luz y que reaccionan químicamente con un sustrato generando un intermediario en estado excitado, que luego vuelve a su estado basal emitiendo luz. La cantidad de luz producida es directamente proporcional a la cantidad de analito presente en la muestra y se determina mediante una curva de calibración a partir de estándares de concentración de Ac conocidos3,13.
Esta técnica posee ventajas con respecto a otras técnicas de detección como: uso de etiquetas no isotópicas, sensibilidad mejorada, rapidez, rango de medición más amplio, aplicabilidad a distintos analitos3,13.
Inmunodifusión radial simple
En una matriz de gel de agarosa con antisuero monoespecífico contra la proteína a determinar, distribuido de manera uniforme, el antígeno (Ag) se difunde libremente, de manera radial, hasta alcanzar la concentración de Ag-Ac adecuada, precipitando en forma de anillo. El cuadrado del diámetro del anillo de precipitación es directamente proporcional a la concentración de Ag. De esta manera, esta técnica permite cuantificar IgG, IgA e IgM mediante una curva de calibración con patrones de concentración para cada Ag13,15.
En la actualidad, ha sido desplazada por otras técnicas debido a que la precipitación completa lleva un tiempo mayor (de 24 a 48 h de difusión en cámara húmeda), y, si existen Igs poliméricas o incompletas, se formarán diferentes halos de precipitación según su peso molecular, que pueden inducir a resultados erróneos13,15.
Inmunoelectroforesis
Esta técnica permite la separación de las Igs en un gel de agar, de acuerdo con la densidad de cargas eléctricas, por exposición a una diferencia de potencial. Luego reaccionan con un antisuero general o específico en una canaleta. Se produce la reacción Ag-Ac, que se visualiza mediante arcos de precipitación que se deben comparar con un patrón conocido2,13,14.
Esta técnica fue reemplazada por la inmunofijación para detectar Igs monoclonales, ya que es poco sensible, lleva un tiempo de 24 h de difusión en cámara húmeda y resulta difícil de interpretar debido a la dificultad para observar la pérdida de asimetría y grosor de las bandas2,7,14.
Causas de hipergammaglobulinemia policlonal
Reactivas
La Hγg policlonal está presente principalmente en infecciones crónicas por virus con gran replicación, persistencia, latencia y con tropismo por células linfoides. Se cree que las diversas especificidades de IgG provocadas por la infección viral resultan del cambio de clase de IgM producida en el repertorio natural de LB16.
La familia de los Herpesviridiae corresponde a este tipo de virus e incluye el citomegalovirus (CMV), el virus del herpes simple (HSV) y el de Epstein Barr (VEB). Estos virus son contraídos principalmente durante la infancia y son prevalentes en pacientes inmunodeprimidos con cuadros clínicos más graves17,18.
Virus del herpes simple
El HSV causa infecciones líticas en la mayoría de las células, persistentes en linfocitos y monocitos e infecciones latentes en neuronas17. Durante la infección herpética, se desarrolla una respuesta humoral dirigida contra las distintas glicoproteínas de envoltura del virus; de esta manera, se produce la Hγg policlonal19.
Virus de Epstein Barr
El VEB es el agente etiológico causante de la mononucleosis infecciosa. Este virus tiene un fuerte tropismo por LB, se acumula en los ganglios linfáticos y persiste en estado latente. Por otro lado, se activan células T en respuesta a los LB infectados para controlar su proliferación3,20. Durante la etapa aguda de la enfermedad se generan diversos Acs inespecíficos transitorios que provocan Hγg policlonal debido a la gran proliferación policlonal de LB y la escasa respuesta celular del huésped de esta fase. Luego, se producen Acs específicos contra distintas proteínas virales que sirven como marcadores del estadio de la infección3,4,20.
Por último, la Hγg es estimulada por la producción de un homólogo de la interleuquina IL-10 por acción del VEB. Este estimula la maduración de los LB y la producción de Acs14.
Citomegalovirus
El CMV infecta células epiteliales y puede permanecer latente en monocitos y linfocitos. La primoinfección por CMV es subclínica y, en ocasiones, similar a la mononucleosis por EBV14,17,20.
La respuesta de Acs contra distintas proteínas del virión lleva a una Hγg policlonal, sin embargo, los Acs neutralizantes tipo IgM e IgG se dirigen principalmente a las glucoproteínas de la cubierta14.
Autoinmunes
Dentro de las patologías autoinmunes, la hepatitis autoinmune (HAI), el síndrome de Sjögren (SSj) y el lupus eritematoso sistémico (LES) son las que cursan en mayor medida con Hγg debido a que existe una pérdida de tolerancia a autoantígenos; esto produce la hiperactivación de LB y, sumado a la influencia de citocinas proinflamatorias como BAFF e IL-6, se da la hiperproducción de IgG, principalmente8,21.
Hepatitis autoinmune
Es una enfermedad hepática inflamatoria crónica que se produce por una respuesta inmunitaria a patógenos exógenos que reaccionan de forma cruzada con autoantígenos hepáticos similares22,23. En consecuencia, se activan los LB y hay una hiperproducción de Igs, donde se eleva la IgG principalmente con un patrón de puente b-g en un PxE, acompañado de un ligero aumento de IgM22,23. Por otro lado, se activan LT, que atacan el parénquima hepático y contribuyen a una inflamación progresiva crónica24.
En conclusión, la HAI se presenta con Hγg policlonal, que es característica de esta patología, lo que permite diferenciar entre un Dco. definitivo y probable de HAI, ya que, a mayores niveles de IgG, existen más chances de presentar HAI24. Además, a la Hγg dos veces mayor que el límite superior de referencia se la considera como un factor de mal pronóstico y se la asocia con mortalidad del 90% a los 10 años sin tratamiento23.
Síndrome de Sjögren
Es una enfermedad que se presenta con infiltración linfoplasmocitaria de las glándulas de secreción exocrina causando sequedad, dolor y fatiga25,26.
Se presentan autoantígenos por las células epiteliales de las glándulas, y se desencadena una respuesta inmune e hiperactividad policlonal de LB con producción aumentada de Acs principalmente tipo IgG26.
Esta Hγg policlonal se relaciona con una mayor prevalencia de manifestaciones extraglandulares. Además, se asocia con pruebas diagnósticas positivas como gammagrafía parotídea y biopsia de glándula salival y, por último, con la presencia de Acs antinucleocitoplasmáticos (ANA), antiantígenos extraíbles del núcleo (ENA) y factor reumatoideo (FR)26.
Lupus eritematoso sistémico
Es una enfermedad autoinmune crónica, multisistémica y compleja con gran heterogeneidad en su presentación clínica y pronóstico27,28. Su fisiopatogenia está dada por una desregulación de la muerte celular por apoptosis; de este modo, se liberan a la circulación autoantígenos modificados y se produce una respuesta inmune específica donde LB autorreactivos generan gran cantidad de Acs causando Hγg de tipo policlonal con predominio de IgG27,28.
Esta Hγg está relacionada con la diversidad de clases y subtipos de Igs autoinmunes presentes. Dentro de estos, se encuentran los ANA, Acs antiribosomal P, FR, y, en menor medida, Acs antifosfolípidos27.
Errores innatos de inmunidad
Se los define como estados patológicos en los que existe un defecto en algún componente del sistema inmune. Suelen manifestarse en los primeros años de vida con infecciones crónicas o recurrentes29.
Síndrome de hiper IgM
Esta patología es causada por la incapacidad de los LB para completar su activación, maduración y diferenciación para ser CP productoras de Acs30. Debido a distintas mutaciones, se produce un defecto en la ruta CD40L-CD40 o en las enzimas necesarias para la hipermutación somática y cambio de isotipo. Por lo tanto, estos pacientes presentan niveles disminuidos de IgG, IgE, IgA y niveles normales o elevados de IgM, que provocan Hγg policlonal30,31. De esta forma, se ve afectada tanto la inmunidad humoral como la celular debido a la cooperación T-B defectuosa. Además, la respuesta de IgM es defectuosa contra distintos agentes infecciosos, lo cual lleva a mayor susceptibilidad a infecciones respiratorias y gastrointestinales32.
Síndrome de hiper IgE
Se trata de una patología caracterizada por niveles muy elevados de IgE, dermatitis eccematoide, infecciones recurrentes de piel y pulmón y formación de abscesos33,34. Puede producirse por mutaciones en distintos genes que condicionan un déficit en la generación de células Th17 a partir de células T CD4+ y, de esta manera, una polarización de la respuesta inmune a un perfil Th2. Este perfil podría facilitar el cambio de clase de Ig hacia IgE provocando una Hγg33,34.
Causas de hipergammaglobulinemia monoclonal
Las GM son un grupo de enfermedades neoplásicas, caracterizadas por la proliferación descontrolada y la acumulación en MO de un clon de CP, acompañada de la hiperproducción de Ig monoclonal. Son más comunes en personas de edad avanzada5.
El evento iniciador es la traslocación del gen de la cadena pesada de la Ig. Luego, los clones sufren eventos genéticos adicionales y, junto con un microambiente inmunosupresor en MO, permiten la evolución de una condición precursora a otra condición maligna35,36.
Gammapatía monoclonal de significado incierto
Es un trastorno clonal de CP no maligno, sin daño de órgano terminal. Algunos factores de riesgo de progresión a malignidad son: una mayor concentración del pico del componente monoclonal a línea de base, picos monoclonales de IgM o IgA, mayor proporción de CP en MO, presencia de proteinuria de Bence Jones, elevación de la velocidad de eritrosedimentación5,37.
Los pacientes resentan un pico monoclonal no IgM en sangre < 3 g/dl, concentración de proteína M en orina <500 mg/día, < 10% CP en biopsia de MO, ausencia de síntomas y daño en órganos dianas. La Hγg suele ser principalmente tipo IgG37,38.
La medición de proteínas es útil para el seguimiento de los pacientes, ya que un pico monoclonal inferior a 1,5 g/dl, de tipo IgG y con un nivel normal de CLLM presenta menor riesgo de transformación en mieloma múltiple (MM)38.
Mieloma múltiple
Es el tumor de CP más frecuente como consecuencia de varios sucesos oncogénicos de la serie B. Además, la IL-6 desempeña un papel primordial en la proliferación de LB e inhibe su muerte por apoptosis38.
Esta patología suele ser precedida por condiciones precursoras como gammapatía monoclonal de significado incierto (GMSI) o MM latente, las cuales son asintomáticas. Algunos factores de riesgo de progresión son: proteína M elevada, isotipo no IgG, radio CLLM anormal, CP en MO >20%35.
Se define por infiltración de MO con CP >10% y presencia de disfunción orgánica5. Esta se puede evidenciarse por hipercalcemia (>11 mg/dl), insuficiencia renal (creatinina sérica >2 mg/dl), anemia (hb <10 g/dl) o lesiones óseas líticas35.
La Hγg monoclonal tiene gran importancia en el Dco. debido a que forma parte de los criterios diagnósticos de otras GM y permite diferenciarlo de estas5. Además, la determinación de la proteína M se usa para el seguimiento del MM, ya que uno de los criterios de remisión completa es una inmunofijación negativa en suero y orina35.
Macroglobulinemia de Waldenstrom
Es una neoplasia de células B causada por una hiperactivación maligna de un LB posgerminal que se transforma antes del cambio de isotipo. Por esta razón, se caracteriza por infiltración de la MO>10 % e Hγg monoclonal de tipo IgM39.
Dentro del sistema internacional de estadificación para macroglobulinemia de Waldenstrom (MGW), aquellos parámetros que se asocian con mayor riesgo son: edad >65 años, hemoglobina <11.5 g/dl, plaquetas <100.000/ ul, b2 microglobulina >3 mg/dl, concentración de IgM >7 g/dl39.
La Hγg monoclonal por IgM conduce a un aumento de la resistencia vascular, la viscosidad sanguínea y posible aparición de autoinmunidad39.
Amiloidosis de cadenas livianas
Es un trastorno de clones de CP que producen Ig monoclonal, que sufre mal plegamiento y agregación formando el depósito amiloide5.
En la MO de estos pacientes, se detecta un porcentaje bajo de CP y existe una restricción clonal de CLLM, principalmente tipo lambda38.
En el PxE de suero, hay ausencia de pico monoclonal debido que las CLLM son filtradas por riñón, pero se encuentran en la orina como una proteinuria de Bence Jones, que puede provocar toxicidad renal. Por lo tanto, para el Dco.de amiloidosis, se realiza inmunofijación en suero y orina (donde se encuentran fisiológicamente concentradas) para caracterizar la paraproteína y, además, medición de CLLM en suero por nefelometría. Para seguimiento de estos pacientes, se miden CLLM en suero5,38,40.
Conclusiones
La detección y caracterización de la Hγg es importante para detectar la causa que lleva a la hiperactivación de LB y aumento de la producción de Igs. De esta manera, permite contribuir al del paciente, diferenciar entre estados clínicos patológicos y, por lo tanto, determinar el tratamiento más adecuado. En el caso de la Hγg policlonal (causas reactivas, autoinmunes y errores innatos de la inmunidad), el PxE y posterior cuantificación por nefelometría o quimioluminiscencia son suficientes para reconocer y caracterizarla, mientras que, en la Hγg monoclonal, se requieren estudios complementarios como inmunoelectroforesis o inmunofijación para caracterizar la paraproteína. Además, en patologías como HAI y las distintas GM, la Hγg ya está dentro de los criterios de clasificación/diagnóstico, por lo tanto, es indispensable la medición de Igs. Asimismo, en HAI, SSj, GMSI y MGW, una mayor Hγg permite establecer un peor pronóstico. Por último, en GM, tales como GMSI y MM, la Hγg es útil para el seguimiento de los pacientes; en el MM, se mide la proteína M por inmunofijación y, en la amiloidosis de CLLM, se mide CLLM en suero por nefelometría.
De este modo, se destaca el papel fundamental del laboratorio, ya que, mediante una gran variedad de técnicas de laboratorio, tales como PxE, inmunofijación, nefelometría y turbidimetría, complementarias entre sí, se pueden caracterizar las diferentes situaciones clínico-patológicas, las cuales son ampliamente utilizadas en los laboratorios actualmente. Sin embargo, se deben conocer las ventajas y desventajas de estas técnicas para poder elegir la más adecuada según la clínica y el PxE del paciente y poder llegar a un Dco., pronóstico y monitoreo adecuados del mismo.
Referencias bibliográficas
1. Abbas AK, Lichtman AH, Pillai S. Inmunología Celular y Molecular. 8va edición. Barcelona, España: Elsevier; 2015.
2. Fainboim L, Geffner J. Introducción a la Inmunología Humana. 6ta edición. Buenos Aires: panamericana; 2011.
3. Henry JB. El Laboratorio en el Diagnóstico Clínico. 20a Edición. Madrid: Marban; 2004.
4. Beuvon C, Martin M, Baillou C, Roblot P, Puyade M. Etiologies of Polyclonal Hypergammaglobulinemia: A scoping review. Eur J InternMed. 2021;(90):119-21, http://dx.doi.org/10.1016/j.ejim.2021.05.023
5. Faria RMD, Silva ROP. Gamopatiasmonoclonais: critérios diagnósticos e diagnósticos diferenciais. RevBrasHematol E Hemoter 2007;29(1): 17-22, http://dx.doi.org/10.1590/S1516-84842007000100005
6. Boban MJ, de Elías R, Kiener O, Kiener G, Jarmi V, Barzón S. Evaluación de la zona gamma del proteinograma por electroforesis: correspondencia clínico-patológica. Acta Bioquím Clín Latinoam 2017; 51(2): 213-20.
7. Rodríguez Zapata M, Sánchez Martínez L, Ruano Soriano E, Montes Ramírez ML. Protocolo de indicaciones e interpretación clínica de la inmunoelectroforesis de las inmunoglobulinas en la práctica clínica. Med - Programa Form Médica Contin Acreditado 2000;8(25):1303-06, http://dx.doi.org/10.1016/S0304-5412(00)70252-6
8. Lo MS, Zurakowski D, Son MBF, Sundel RP. Hypergammaglobulinemia in the pediatric population as a marker for underlying autoimmune disease: a retrospective cohort study. Pediatr Rheumatol 2013;11(42): 1-8, http://dx.doi.org/10.1186/1546-0096-11-42
9. Paniagua MJP, Pérez RDB. El sistema inmune del recién nacido. Alergia, asma e inmunología pediátricas. Medigraphic 2003;12(2):63-68.
10. Torres LS, Gamboa DÁ. Inmunología Perinatal. FEMINA 2014; 42(4): 185-192.
11. Bastias C, Sidgman F, Rodríguez C. Laboratorio de Inmunología en la práctica clínica. Rev. Med. Clin. Condes 2015;26(6):764-75, http://dx.doi.org/10.1016/j.rmclc.2015.11.005
12. Pérez Surribas D, Cárdenas Fernández MC, Zapico Muñiz D. Recomendaciones sobre la separación electroforética de las proteínas plasmáticas en suero. Documentos de la SEQC 2015; 91-104.
13. González de Buitrago JM. Técnicas y métodos de laboratorio clínico. 3a edición. Barcelona: Elsevier Masson; 2010.
14. Parslow TG, Stites DP, Terr AI, Imboden JB. Inmunología básica y clínica. 10ª Edición. México, D.F: El manual moderno; 2002.
15. Lomonte B, Bonilla C. Castro, E. Niveles de Inmunoglobulinas Séricas (IgG, IgA e IgM) en adultos jóvenes sanos, por el método de inmunodifusión radial. Rev. costarric. cienc. Méd 1985 ;6(4):183-190.
16. Hunziker L, Recher M, Macpherson AJ, Ciurea A, Freigang S, Hengartner H, et al. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nat Immunol 2003;4(4):343-9, http://dx.doi.org/10.1038/ni911
17. Bascones-Martínez A, Pousa-Castro X. Herpesvirus. Av Odontoestomatol 2011; 27(1):11-24.
18. Drouet E, Chapuis-Cellier C, Bosshard S, Verniol C, Niveleau A, Touraine JL, et al. Oligo-monoclonal immunoglobulins frequently develop during concurrent cytomegalovirus (CMV) and Epstein–Barr virus (EBV) infections in patients after renal transplantation. Clin ExpImmunol 2001;118(3):465-72, http://dx.doi.org/10.1046/j.1365-2249.1999.01084.x
19. Hantz S, Alain S. Infecciones por el virus del herpes simple. EMC Pediatría 2018;53(2):1-13. DOI: 10.1016/S1245-1789(18)89722-0
20. Tinoco Racero I, Caro Gómez N, Rodríguez Leal C, López Tinoco E. Infecciones por el virus de Epstein-Barr y citomegalovirus. Medicine 2014;11(50):2954-64, http://dx.doi.org/10.1016/S0304-5412(14)70722-X
21. Ehrenstein MR, Isenberg DA. Hypergammaglobulinaemia and autoimmune rheumatic diseases. Ann RheumDis 1992;51(11):1185-7, http://dx.doi.org/10.1136/ard.51.11.1185
22. Liberal R, Mieli-Vergani G, Vergani D. Autoimmune hepatitis: From mechanism so therapy. Rev Clínica Esp 2016;216(7):372-83. En prensa. http://dx.doi.org/10.1016/j.rce.2016.04.003
23. Orts Costa JA, Zúñiga Cabrera A, Alarcón Torres I. Hepatitis autoinmune. AnMed Interna 2004;21(7):34-48.
24. Benítez-Rodríguez B, Rodríguez-Sicilia MJ, Vázquez-Morón JM, Pallarés-Manrique H, Ramos-Lora M. Hepatitis Autoinmune: etiopatogenia, diagnóstico y tratamiento. RAPD ONLINE 2011;32 (2): 86-93.
25. Nocturne G. Actualitésdans le syndrome de Sjögren primitif : aspectscliniques et thérapeutiques. RevMédecine Interne 2019;40(7):433-9, http://dx.doi.org/10.1016/j.revmed.2019.03.329
26. M. Ramos-Casalsa, M. García-Carrascoa, R. Cerveraa, J. Fonta, M. Ingelmoa. Significado clínico de las alteraciones analíticas en el síndrome de Sjögren. Revista Española de Reumatología 2002;29(7):343-55.
27. Mesa JFC, López AEL, Torres JC. Lupus eritematoso sistémico en actividad y cociente albúmina/globulina invertido, ¿hallazgo propio de la enfermedad? RCuR 2020; 22(Suppl 1): e853
28. Mok MY. The immunological basis of B-cell therapy in systemic lupus erythematosus. Int J RheumDis 2010;13(1):3-11, http://dx.doi.org/10.1111/j.1756-185X.2009.01458.x.
29. Zamora G, Victoria M. Inmunodeficiencias en la infancia. Rev Cubana Pediatr [Internet] 2003 [citado 2024 Ene 09]; 75(4). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312003000400007&lng=es.
30. Jesus AA, Duarte AJS, Oliveira JB. Autoimmunity in Hyper-IgM Syndrome. J Clin Immunol 2008;28(S1):62-6, http://dx.doi.org/10.1007/s10875-008-9171-x
31. Zhao, EJ, Cheng, CV, Mattman, A, Chen, LY. Polyclonal hypergammaglobulinemia: assessment, clinical interpretation, and management. The Lancet Haematology 2021;8(5):e365-e375, https://doi.org/10.1016/S1470-2045(23)00223-1
32. França TT, Barreiros LA, al-Ramadi BK, Ochs HD, Cabral-Marques O, Condino-Neto A. CD40 ligand deficiency: treatment strategies and novel therapeutic perspectives. Expert Rev Clin Immunol 2019;15(5):529-40, http://dx.doi.org/10.1080/1744666X.2019.1573674.
33. Guisado Vasco P, Fraile Rodríguez G, Arechaga Uriarte S. Síndrome de hipergammaglobulinemia IgE con infecciones recurrentes: patogénesis, diagnóstico y aproximación terapéutica. Rev Clínica Esp 2011;211(10):520-6, http://dx.doi.org/10.1016/j.rce.2011.04.009
34. Tagle C MT, Melys G A, Castillo M A, Norambuena R X, Quezada L A. Síndrome Hiper IgE: a propósito de tres casos clínicos. RevChil Pediatría 2014;85(3):328-336.
35. van de Donk NWCJ, Pawlyn C, Yong KL. Multiple myeloma. The Lancet 2021;397:410-427, http://dx.doi.org/10.4067/S0370-41062014000300009
36. Ho M, Patel A, Goh CY, Moscvin M, Zhang L, Bianchi G. Changing paradigms in diagnosis and treatment of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Leukemia 2020;34(12):3111-3125, http://dx.doi.org/10.1038/s41375-020-01051-x
37. Amaador K, Peeters H, Minnema MC, Nguyen TQ, Dendooven A, Vos JMI, et al. Monoclonal gammopathy of renal significance (MGRS): histopathologic classification, diagnostic workup, and therapeutic options. Neth J Med 2019;77(07): 243-254.
38. Charlot-Lambrecht I, Salmon JH, Gagneux-Lemoussu L, Brochot P, Eschard JP. Mieloma múltiple. EMC - AparLocomot 2012;45(1):1-13, http://dx.doi.org/10.1016/S1286-935X(12)60820-X
39. Gertz MA. Waldenström macroglobulinemia: 2023 update on diagnosis, risk stratification, and management. Am J Hematol 2023; 98(2):348-358, http://dx.doi.org/10.1002/ajh.26796.
40. Yadav P, Leung N, Sanders PW, Cockwell P. The use of immunoglobulin light chain assays in the diagnosis of paraprotein-related kidney disease. Kidney Int 2015;87(4):692-7, http://dx.doi.org/10.1038/ki.2014.333
Notas
Notas de autor
luciacastellano3@gmail.com
Información adicional
redalyc-journal-id: 651