Artículos originales
Recepción: 25 Julio 2016
Aprobación: 19 Abril 2017
DOI: 10.7705/biomedica.v38i0.3454
Resumen
Introducción.: La enfermedad por almacenamiento de glucógeno de tipo III es una alteración autosómica recesiva, en la cual las mutaciones del gen AGL causan una deficiencia en la enzima desramificadora de glucógeno. Se caracteriza por hipoglucemia, hepatomegalia y miopatías progresivas. El análisis molecular del gen AGL ha evidenciado mutaciones que difieren según la población estudiada. En la actualidad, no existen reportes que describan mutaciones en el AGL de pacientes colombianos con esta condición.
Objetivo.: Describir las características clínicas y moleculares de diez pacientes colombianos con enfermedad por almacenamiento del glucógeno de tipo III.
Materiales y métodos.: Se analizaron diez pacientes pediátricos colombianos con la enfermedad y se hizo su estudio genético mediante la secuenciación de las regiones que codifican y las intrónicas circundantes del gen AGL con el método de Sanger.
Resultados.: Todos los pacientes tenían el fenotipo clásico de la enfermedad. El estudio genético reveló la mutación p.Arg910X en dos pacientes. Uno presentó la mutación p.Glu1072AspfsX36 y otro resultó heterocigoto compuesto con las mutaciones p.Arg910X y p.Glu1072AspfsX36. Asimismo, en tres pacientes se detectó la deleción de los exones 4, 5 y 6 del gen AGL. Los estudios de simulación computacional predijeron que estos defectos eran patogénicos. En tres pacientes no se encontraron mutaciones en las regiones amplificadas.
Conclusión.: Se encontraron mutaciones y deleciones que explican el fenotipo clínico de los pacientes. Este es el primer reporte en el que se describe el fenotipo clínico y el espectro de mutaciones en el gen AGL de pacientes colombianos, lo cual es importante para ofrecer un apropiado pronóstico, y asesoría genética al paciente y a su familia.
Palabras clave: enfermedad del almacenamiento de glucógeno de tipo III/diagnóstico, glucógeno, sistema de la enzima desramificadora del glucógeno, glucogenólisis..
Abstract
Introduction: Type III glycogen storage disease (GSD III) is an autosomal recessive disorder in which a mutation in the AGL gene causes deficiency of the glycogen debranching enzyme. The disease is characterized by fasting hypoglycemia, hepatomegaly and progressive myopathy. Molecular analyses of AGL have indicated heterogeneity depending on ethnic groups. The full spectrum of AGL mutations in Colombia remains unclear.
Objective: To describe the clinical and molecular characteristics of ten Colombian patients diagnosed with GSD III.
Materials and methods: We recruited ten Colombian children with a clinical and biochemical diagnosis of GSD III to undergo genetic testing. The full coding exons and the relevant exon-intron boundaries of the AGL underwent Sanger sequencing to identify mutation.
Results: All patients had the classic phenotype of the GSD III. Genetic analysis revealed a mutation p.Arg910X in two patients. One patient had the mutation p.Glu1072AspfsX36, and one case showed a compound heterozygosity with p.Arg910X and p.Glu1072AspfsX36 mutations. We also detected the deletion of AGL gene 3, 4, 5, and 6 exons in three patients. The in silico studies predicted that these defects are pathogenic. No mutations were detected in the amplified regions in three patients.
Conclusion: We found mutations and deletions that explain the clinical phenotype of GSDIII patients. This is the first report with a description of the clinical phenotype and the spectrum of AGLmutations in Colombian patients. This is importantto provide appropriate prognosis and genetic counseling to the patient and their relatives.
Key words: esENGlycogen storage disease type III/diagnosis, glycogen, glycogen debranching enzyme system, glycogenolysis.
La enfermedad por almacenamiento del glucógeno de tipo III (glucogenosis de tipo III; #232400 en la base de datos de Online Mendelian Inheritance in Man ® ,OMIM), también conocida como enfermedad de Cori o de Forbes, es un trastorno metabólico autosómico recesivo causado por mutaciones en el gen AGL que codifica la enzima desramificadora del glucógeno. Esta enzima, de 1.532 aminoácidos, tiene dos actividades catalíticas independientes: amilo-1,6-glucosidasa (EC3.2.1.33) y oligo-1,4-1,4-glucantransferasa (EC2.4.1.25), y cataliza el último paso en la conversión de glucógeno en glucosa1-7. Por ello, su deficiencia provoca acumulación de una forma intermedia del glucógeno, la dextrina límite, en los órganos afectados (hígado, músculo esquelético y músculo cardiaco), y su disfunción secundaria8,9.
Existen diferentes subtipos de la enfermedad en función de la pérdida de la actividad de esta enzima en el hígado o en los músculos, lo cual da lugar a diversos fenotipos clínicos con características heterogéneas10. La enfermedad representa, aproximadamente, el 24 % de todos los casos de glucogenosis y su incidenciaestimada en Europa es de un caso por cada 83.000 nacidos vivos, y de uno por cada 100.000 nacidos vivos en Norte-américa2,8,11. En poblaciones especiales, como la de las Islas Feroe y la comunidad judía del norte de África, la prevalencia es de 1:3.600 y 1:5.400, respectivamente11,12.
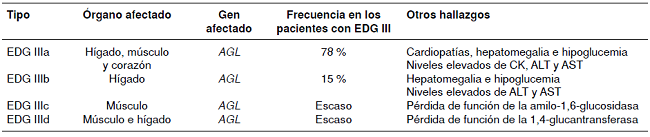
La enfermedad por almacenamiento del glucógeno de tipo IIIa es el subtipo más común (78 %), y afecta el hígado, el músculo esquelético y el cardíaco; el subtipo IIIb afecta a 15 % de los pacientes e involucra solo al hígado13,14. Esporádicamente, se han detectadocasos con pérdida selectiva de la actividad de la glucosidasa (subtipo IIIc) o de la actividad de la transferasa (subtipo IIId)14-16. Las características clínicas y moleculares más importantes de los subtipos de la enfermedad se presentan en el cuadro suplementario S1.
En la infancia, las principales características clínicas de esta enfermedad son la hipoglucemia, la hepatomegalia, las miopatías esqueléticas y las cardiomiopatías. En los análisis bioquímicos, se encuentra hipoglucemia desencadenada por el ayuno y aumento de los niveles séricos de triglicéridos, colesterol, urea, creatina fosfocinasa (CPK), aspartato aminotransferasa (AST) y alanino aminotransferasa (ALT).
El genAGL está localizado en el cromosoma 1p21.2 y consta de 35 exones que incluyen una secuencia genómica de 85 kb17-19. Hasta la fecha, se han descrito más de 100 mutaciones del gen AGL, especialmente en poblaciones del norte de Europa y de Egipto, así como en hispanos y asiáticos4-6,9,14,20-31.
Actualmente, el diagnóstico definitivo de la enfermedad se establece mediante estudios moleculares no invasivos que detectan las mutaciones del genAGL en pacientes con sospecha clínica y bioquímica de la enfermedad. Esta tecnología ha desplazado los métodos utilizados en el pasado, como la biopsia hepáticay la de músculo, con las cuales se evidencia la acumulación de glucógeno anormal (dextrina límite) en los hepatocitos y las alteraciones histológicas musculares8,26.
Hasta el momento no existe un tratamiento específico para las glucogenosis; sin embargo,la alimentación nasogástrica u oral nocturna frecuente, el suministro de maltodextrina (almidón de maíz) cruda y la alimentación a intervalos cortos de tiempo con restricción de sacarosa, ayudan a controlar la hipoglucemia y la hepatomegalia. Este tipo de tratamientos también restaura el crecimiento de los niños afectados, y controla las alteraciones bioquímicas, lo cual mejora la función del músculo esquelético y el cardiaco8.
No hay estadísticas claras y completas sobre la incidencia y el espectro de mutaciones del gen AGL relacionadas con esta enfermedad en Latinoamérica ni en la población colombiana.
En este contexto, el objetivo del presente estudio fue describir el fenotipo clínico e identificar las bases moleculares de la enfermedad por acumulación del glucógeno de tipo III en un grupo de diez niños colombianos con diagnóstico clínico, bioquímico e histológico de la enfermedad.
Materiales y métodos
Población de estudio
El estudio se hizo en diez pacientes pediátricos (seis niñas y cuatro niños) de diez familias no relacionadas, provenientes de varias regiones de Colombia, pero principalmente de Antioquia, con diagnóstico previo indicativo de la enfermedad, establecido con base en sus manifestaciones clínicas, el perfil bioquímico y el estudio histológico.
Todos los pacientes fueron diagnosticados y tratados en el Hospital Pablo Tobón Uribe de Medellín. Las pruebas de laboratorio clínico para determinar el diagnóstico bioquímico, incluyeron la determinación en suero de ALT, AST, bilirrubina total, bilirrubina directa, gamma glutamiltranspeptidasa (GGT), fosfatasa alcalina, triglicéridos, colesterol total, ácido úrico, creatina fosfocinasa total (CPK) y la fracción MB (CPK-MB), y glucemia en ayuno, por lo menos, de cuatro horas.
Los pacientes presentaron efectos hepáticos, musculares y cardiacos evidenciados por las alteraciones bioquímicas, y mediante ecografía abdominal y ecocardiografía. La biopsia de tejido hepático se hizo mediante punción dirigida por ecografía. En el estudio histopatológico se utilizaron las tinciones de hematoxilina y eosina, ácido peryódico de Schiff (PAS) y tricrómica, para evidenciar la acumulación de glucógeno en los hepatocitos y la fibrosis portal con formación de bandas delgadas de tejido conjuntivo.
Extracción del ADN de sangre periférica
Se obtuvo una muestra de sangre periférica por punción venosa de los pacientes del estudio y de un control sano adulto, utilizando tubos de 5 ml con suplemento de heparina sódica (BD Vacutainer). Para la extracción del ADN genómico (ADNg) a partir de las muestras de sangre, se utilizó un estuche comercial (QIAamp DNA Blood Mini Kit™, Qiagen, USA).
La pureza y la concentración del ADNg obtenido se estimaron mediante espectrofotometría, utilizando el equipo NanoDrop 2000™ (Thermo Fisher Scientific, USA). La integridad y la cantidad de ADNg se analizaron en un gel de agarosa al 1 % y mediante espectrofotometría, respectivamente.
Reacción en cadena de la polimerasa y secuenciación
Con el objetivo de detectar mutaciones del genAGL, los exones 3 al 35 y las regiones intrónicas circundantes (aproximadamente 50 pb en la región 5’ y 3’ del exón) se amplificaron mediante reacción en cadena de la polimerasa (PCR), utilizando ADNg como molde.
Se utilizó el estuche comercial PCR Master Mix 2X™ (Thermo Fisher Scientific, USA) en una reacción con un volumen final de 30 µl que contenía 150 ng de ADNg y 0,3 µM de cada uno de los oligonucleótidos. Para todas las reacciones, se utilizaron los oligonucleótidos y las condiciones de la PCR descritas previamente por Goldstein, et al. 26. Los oligonucleótidos y ciclos usados en cada una de las PCR se describen en los cuadro suplementario 2S y cuadro suplementario 3S.
Todos los productos de PCR se purificaron y secuenciaron utilizando un secuenciador automático Applied Biosystems ABI 3730™ (Thermo Fisher Scientific, USA) con los oligonucleótidos de sentido y antisentido utilizadospara cada una de las reacciones de PCR.
Los cromatogramas obtenidos se analizaron con el programa Vector NTI™ (Invitrogen), utilizando como referencia la secuencia de la isoforma 1 del genAGL reportada en la base de datos delNational Center for Biotechnology Information (NCBI) (NM_000028.2).
La posición de las variantes se determinó mediante el programa Alamut Visual™ (Interactive Biosoftware, Francia), versión 2.7.2, utilizando el genoma de referencia GRCh38, en tanto que el análisis de la patogenia de las mutaciones encontradas se hizo con los programas SIFT, PolyPhen y CADD32-34.
La nomenclatura utilizada para nombrar las variantes y las mutaciones, se basó en las guías de nomenclatura de la Human Genome Variation Society (HGVS)35,36.
Matriz enfocada en exones basada en la hibridación genómica comparativa
Con el objetivo de identificar en una escala de alta resolución la ganancia o la pérdida de cromosomas completos o regiones subcromosómicas y cambios del número de copias en una resolución de exones individuales en la región 1p21.2 del cromosoma 1, se hizo un ensayo utilizando una matrizenfocada en el genAGLde hibridación genómicacomparativa basada en exones (Exome Array Comparative Genomic Hybridization - Exon ArrayCGH). El ensayo se hizo en la empresa GeneDx (GeneDx, Gaithersburg, Maryland, USA), siguiendo todas las recomendaciones técnicas sugeridas37,38.
Consideraciones éticas
Los padres de los pacientes diligenciaron y firmaron el consentimiento informado para el estudio molecular según las recomendaciones y la aprobación previa del Comité de Bioética para Investigación en Humanos de la Universidad de Antioquia y del Hospital Pablo Tobón Uribe. Asimismo, el estudio fue revisado por el Comité de Bioética para Investigación en Humanos de la Universidad de Antioquia y aprobado como investigación “sin ningún riesgo” según los lineamientos de la Declaración de Helsinki.
Resultados
Resultados clínicos y bioquímicos
La población de estudio estaba conformada por pacientes pediátricos de sexo femenino (n=6) y masculino (n=4) que, en el momento del diagnóstico de la enfermedad por almacenamiento del glucógeno de tipo III, tenían un promedio de edad de 38 meses (rango de 5 meses a 12 años). En el cuadro 1 se muestran las características clínicas de cada uno de los pacientes, en las cuales predomina la hepatomegalia y las cardiopatías (100 % de los casos).
Características clínicas y demográficas de los pacientes con diagnóstico de enfermedad por almacenamiento delglucógeno de tipo III
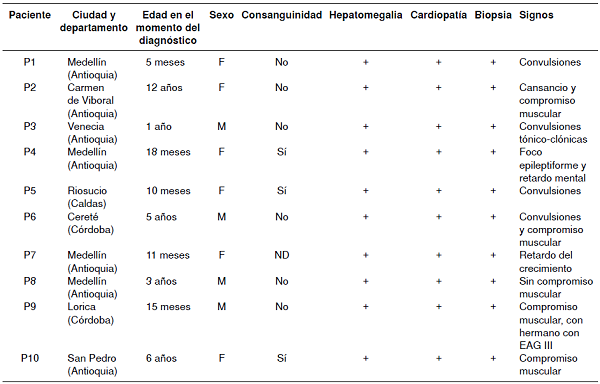
Los niños pertenecían a diez familias no relacionadas, provenientes de varias regiones de Colombia: siete de Antioquia (zona centro y noroccidental de Colombia), dos de Córdoba (zona norte de Colombia) y uno de Caldas (zona centro de Colombia). Los padres de tres de los pacientes (P4, P5 y P10) eran consanguíneos.
En la biopsia de tejido hepático de todos los pacientes, se observaron hepatocitos agrandados con abundante material intracelular, positivo con el uso de PAS, correspondiente a glucógeno, así como fibrosis de varios grados, lo cual es un signo complementario de la enfermedad (figura 1).
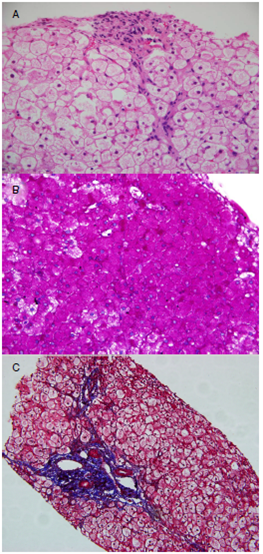
Figura 1
Estudio histopatológico de tejido hepático obtenido depacientes con enfermedad por almacenamiento del glucógenode tipo III. A. Hepatocitos con membrana celular bien definida,citoplasma amplio, claro, cargado de glucógeno y núcleos redondeados en forma de haba (célula vegetal), desplazados hacia laperiferia en biopsia hepática. Hematoxilina y eosina, 400X. B.Hepatocitos con abundante glucógeno citoplásmico en biopsiahepática. Ácido peryódico de Schiff (PAS), 400X. C. Fibrosisportal con formación de bandas delgadas de tejido conjuntivoen biopsia hepática. Tinción tricrómica de Masson, 400X.
En los análisis bioquímicos en suero de los parámetros de laboratorio, se evidenció un incremento de la concentración de CPK y de CPK-MB en 70 y 90 % de los casos, respectivamente (cuadro 2). Se encontró, asimismo, un incremento en la concentración sérica de la ALT, con un valor promedio de 457 UI/L, y de la AST, con un valor promedio de 488 UI/L en el 100 % de los casos, aunque se pudieron evidenciar diferencias entre los pacientes según el momento en que se hizo el estudio bioquímico.
Características bioquímicas de los pacientes con diagnóstico de enfermedad por el almacenamiento del glucógeno detipo III
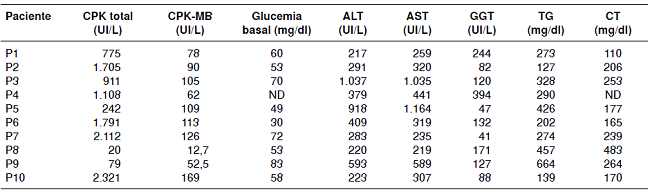
En 60 % de los casos se encontraron valores de glucosa menores de 70 mg/dl (promedio: 59 mg/dl), lo cual corresponde a hipoglucemia.
Por último, se evidenciaron valores de triglicéridos superiores a 200 mg/dl (valor promedio: 318 mg/dl) en 80 % de los casos y, de colesterol total superiores a 240 mg/dl, en 30 % de los pacientes estudiados (valor promedio: 229 mg/dl).
Análisis de mutaciones
Como se observa en el cuadro 3, se identificaron varios tipos de mutaciones del ADNg de los pacientes. Una de ellas, la mutación c.2728C>T en el exón 22 donde se genera un codón de terminación prematuro (p.Arg910X), se detectó en los pacientes P2 y P5 en condición homocigota. El paciente P5 es hijo de padres consanguíneos (figura 2a y cuadro 3). Del mismo modo, se encontró la deleción c.3216_3217delGA en un paciente hijo de padres consanguíneos (caso P4). Esta deleción homocigótica detectada en el exón 25, induce un cambio en el marco de lectura a partir del codón 1072 y, 36 codones corriente abajo, se origina un codón prematuro de terminación (p.Glu1072AspfsX36) (figura 2b y cuadro 3). El paciente P3 es un heterocigótico compuesto, lo que no se había reportado previamente en la literatura científica, ya que posee el cambio c.2728C>T en el exón 22 en estado heterocigótico y es igualmente heterocigótico para la deleción c.3216_3217delGA en el exón 25 (figura 2c y cuadro 3), alelos no funcionales que explican el fenotipo del paciente. Los exones 22 y 25 del gen AGL se secuenciaron en los padres del paciente: en el padre solo se encontró el cambio c.2728C>T y, en la madre, solo la deleción c.3216_3217delGA, en ambos casos en estado heterocigótico.
Mutaciones del gen AGL caracterizadas en muestras de pacientes con diagnóstico de enfermedad por almacenamientodel glucógeno de tipo III

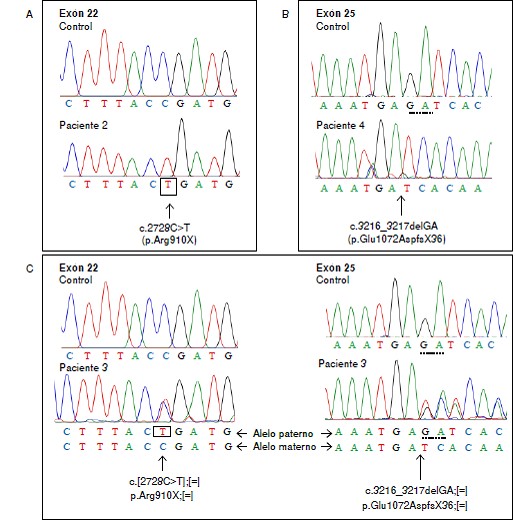
Figura 2
Análisis del gen AGL mediante cromatograma de las secuencias correspondientes al control sano y a los pacientes P2, P3 y P4. A. Cromatogramas del exón 22 correspondientes al control y al paciente P2. El cambio homocigótico c.2728C>T se indica con un recuadro en el nucleótido afectado. B. Cromatogramas del exón 25 del control y del paciente P4; los nucleótidos GA se señalan en la secuencia del control con una línea punteada que indica los nucleótidos que se pierden en la deleción homocigótica c.3216_3217delGA en este paciente; en la secuencia se señala con una flecha el lugar donde ocurrió la deleción y se inicia el cambio en el marco de lectura. C. Se muestran los cromatogramas de los exones 22 (izquierda) y 25 (derecha) del paciente P3 y del control. En el exón 22, el cambio heterocigótico c.2728C>T se indica con un recuadro en el alelo paterno, mientras que, en la misma posición en el alelo materno , se conserva el nucleótido de tipo silvestre. En la secuencia del exón 25, se señalan los nucleótidos GA con una línea punteada en la secuencia del control y el alelo paterno del paciente, indicando los nucleótidos que se pierden en estado heterocigótico en el paciente para la deleción c.3216_3217delGA . En el alelo materno de la secuencia del paciente, se señala con una flecha el lugar donde ocurrió la deleción y se inicia el cambio en el marco de lectura. La posición de los nucleótidos se basa en la secuencia de ADNg reportada en el National Center for Biotechnology Information (NCBI) con el código NM_000028.2 y el genoma de referencia GRCh38
Los exones 4, 5 y 6 del gen AGL no se pudieron amplificar en las muestras provenientes de los casos P1, P7 y P10, aun cuando con el mismo protocolo estos exones amplificaron en las muestras del control sano y de los otros pacientes del estudio. En la figura 3 se presenta un gel representativo de los productos de PCR del exón 4 de los pacientes y del control; en este caso no se observan amplicones en los pacientes P1, P7 y P10. Estos resultados se repitieron al analizar los exones 5 y 6 del gen AGL, lo cual sugiere que estos pacientes poseen una deleción que incluye los exones 4, 5 y 6 y sus regiones intrónicas; los exones restantes de estos pacientes se amplificaron normalmente mediante PCR. Este resultado se corroboró en el paciente P1 mediante hibridación genómica comparativa, con la cual se determinó la ausencia de los exones 4, 5 y 6 (figura 4).
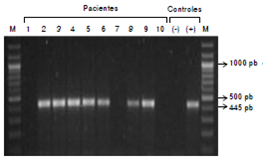
Figura 3
Electroforesis de productos de PCR del exón 4 del genAGL. Dicho exón se amplificó mediante PCR a partir de ADNgenómico en los diez pacientes estudiados y en el individuosano que sirvió como control positivo (+). Posteriormente, serealizó una electroforesis en gel de agarosa al 2 % utilizandocada uno de los productos de la PCR y se determinó, mediantela tinción con bromuro de etidio, la presencia de una bandaequivalente a 445 pb. Para el control negativo de la PCR (-),se utilizaron todos los reactivos de la PCR excepto el ADNg.
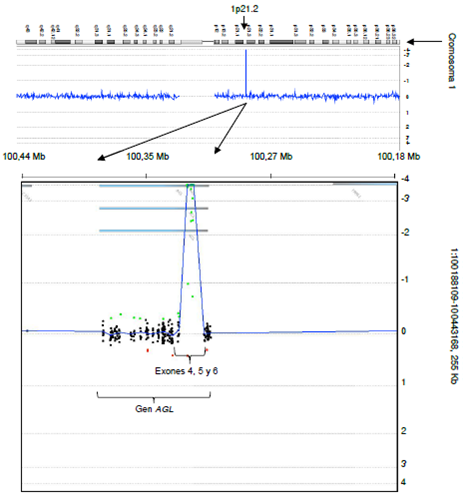
Figura 4
Hibridación genómica comparativa (aCGH) enfocada en exones. El ensayo, enfocado en la región 1p21.2 del cromosoma1, indicó que el paciente P1 es homocigótico para una deleción que incluye los exones 4, 5 y 6 del gen AGL. Asimismo, se observóun número de copias normales en el resto de los exones evaluados en este mismo gen. El log ratio se observa en el gráfico(intensidad log2 del fluorocromo en el eje de la X). Las deleciones se observan como desviaciones negativas con respecto al logratio (puntos verdes)
Por último, en los pacientes P6, P8 y P9 no se encontraron mutaciones en los 35 exones y las regiones intrónicas próximas bajo estudio, por lo cual no se pudo ofrecer una explicación molecular para su fenotipo clínico.
Identificación de polimorfismos
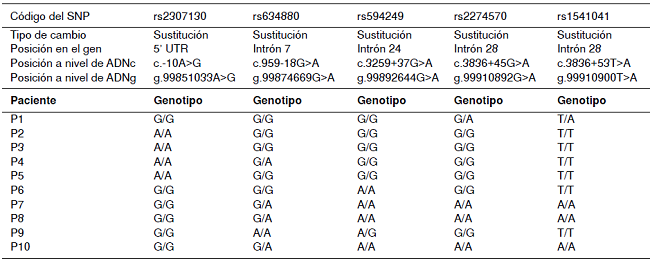
Durante la secuenciación del genAGL, se identificaron cinco polimorfismos de un solo nucleótido (Single Nucleotide Polymorphism, SNP) muy frecuentes en la población, ya reportados en la literatura y que no están asociados al fenotipo clínico de la enfermedad por almacenamiento del glucógeno de tipo III. Estos SNP son: c.-10A>G SNP (código dbSNP rs2307130), c.959-18G>A (rs634880), c.3259+37G>A (rs594249), c.3836+45G>A (rs2274570) y c.3836+53T>A (rs154104) (cuadro 4).
Polimorfismos encontrados en pacientes colombianos con diagnóstico de glucogenosis de tipo III
Discusión
El diagnóstico de la enfermedad por almacenamiento de glucógeno de tipo III exige la combinación de estudios clínicos, bioquímicos y moleculares. Actualmente, el estudio molecular de la enfermedad se facilita gracias a la clonación y a la caracterización molecular del genAGL,que han permitido identificar sus características. En este estudio se exploró la presencia de mutaciones del gen AGLde diez pacientes pediátricos colombianos con diagnóstico de esta condición, establecido con base en sus manifestaciones clínicas, el perfil bioquímico y el estudio histológico.
En los casos estudiados predominaron las características clínicas y bioquímicas propias de la enfermedad: hipoglucemia, hepatomegalia, miocardiopatía, y aumento de las transaminasas y la CPK, como se ha reportado ampliamente en la literatura8,39. Estos signos clínicos se atribuyen a la degradación anormal y a la acumulación de glucógeno, lo cual produce disfunción hepática (hipoglucemia, hepatomegalia pronunciada y, en algunos casos, cirrosis), miopatía esquelética, cardiomiopatía y crecimiento deficiente40.
Las cardiomiopatías, con hallazgo de miocardiopatía hipertrófica en la ecocardiografía, ocurren en la mayoría de los individuos con el subtipo IIIa de la enfermedad y, frecuentemente, aparecen durante la infancia39.
La hipoglucemia es una manifestación clínica temprana de la enfermedad que se debe a un defecto en la glucogenólisis debido a la deficiencia en la actividad de la enzima desramificadora de glucógeno, lo cual conduce a que el glucógeno almacenado en el hígado no pueda ser utilizado para la homeostasis de la glucosa, y resulta en una hipoglucemia significativa, especialmente después del ayuno.
En contraste con otros reportes de pacientes con esta condición, en este estudio se encontró que la hipoglucemia estaba presente en el 60 % de los pacientes39,40, lo cual concuerda con un reciente estudio de Sentner, et al.8, quienes reportaron hipoglucemia en la mitad de los pacientes, lo que, según los autores, demuestra que, en pacientes con hepatomegalia, transaminasas elevadas e hiperlipidemia, no se debe obviar el diagnóstico de la enfermedad por almacenamiento del glucógeno de tipo III en ausencia de hipoglucemia grave8.
En general, en este estudio se evidenció un incremento de la concentración sérica de las enzimas CPK, CPK-MB, ALT y AST, lo cual se correlaciona con la afectación del músculo cardíaco y el esquelético, y con la citólisis causada por la acumulación anormal de glucógeno en los hepatocitos, como se constató en todos los participantes del presente estudio y en otros consignados en la literatura científica8,41,42. También, fue evidente el incremento de los lípidos séricos al detectarse hipertrigliceridemia e hipercolesterolemia en 80 y 30 % de los casos, respectivamente, lo que concuerda con estudios previos en los cuales se reporta que la concentración sérica de los triglicéridos en pacientes con esta enfermedad puede fluctuar entre 200 y 500 mg/dl39, así como con los datos reportados en elInternational Study on Glycogen Storage Disease (ISGSDIII) que dan cuenta de concentraciones elevadas de colesterol en 34 % y de triglicéridos en 72 a 79 % de los casos estudiados8.
Aunque los parámetros clínicos y bioquímicos descritos ayudan al diagnóstico de la enfermedad, estos no son específicos y podrían presentarse en otras enfermedades metabólicas, especialmente la glucogenosis de tipo I. Por ello, el análisis genético es determinante para el diagnóstico de los pacientes con sospecha de esta enfermedad8,39.
En este estudio no se investigó la actividad de la enzima desramificadora del glucógeno en ninguno de los pacientes; sin embargo, para el diagnóstico y manejo de los pacientes, se hicieron biopsias del tejido hepático, las cuales demostraron hepatocitos con abundante material intracelular positivo para PAS correspondiente a glucógeno y fibrosis en el 100 % de los pacientes, resultados que permitieron confirmar el diagnóstico8.
En este contexto, en el presente estudio se hizo el análisis genético de los pacientes cuyas características clínicas, parámetros bioquímicos y estudios histológicos correspondían al diagnóstico de la enfermedad. En el caso de los pacientes P2 y P5, se encontró la mutación sin sentido c.2728C>T en estado homocigótico, la cual induce el cambio del aminoácido arginina en la posición 910 por un codón de terminación (p.Arg910X), generándose así una proteína truncada que pierde el dominio amilo-1,6-glucosidasa y el dominio de unión al glucógeno, y se clasificó como deletérea y patogénica según el análisis con los programas SIFT, CADD y Polyphen.
Esta mutación, parte de un compuesto heterocigoto con la mutación c.753_756delGACA, se reportó inicialmente en Italia, en una paciente de tres años de edad diagnosticada con la condición, cuyo caso era similar al de los pacientes colombianos desde el punto de vista clínico, excepto que no presentó compromiso cardiaco43. En el 2016, en un estudio multicéntrico que incluía 175 pacientes con esta enfermedad provenientes de diez países, Sentner, et al., reportaron nuevamente el cambio p.Arg910X, sin embargo, no aportaron datos adicionales sobre la frecuencia, el origen y las características clínicas específicas del hallazgo8.
En el presente estudio, se detectó en el paciente P4, hijo de padres consanguíneos, la mutación homocigótica c.3216_3217delGA, la cual induce un cambio en el marco de lectura, con la posterior aparición de un codón de parada prematuro (p.Glu1072AspfsX36) en que la proteína pierde el dominio amilo-1,6-glucosidasa y el de unión al glucógeno, con efectos deletéreos evidenciados por los predictores utilizados. Dicha mutación se reportó en el estudio de Goldstein, et al., en 201026, en un paciente de origen desconocido incluido en una cohorte multiétnica de 25 pacientes con la enfermedad26. Asimismo, Sentner, et al., la reportaron nuevamente en un paciente de origen desconocido8.
Con respecto al paciente P3 de este estudio, es importante mencionar que, hasta donde se sabe, es la primera vez que se reporta en el mundo una condición de compuesto heterocigótico para los cambios patogénicos p.Arg910X y p.Glu1072AspfsX36 en un caso de enfermedad por almacenamiento del glucógeno de tipo III.
En este estudio se identificó también la deleción de una región del cromosoma 1p21.2 que comprende los exones 4, 5 y 6 del gen AGL,en dos pacientes hijos de padres no consanguíneos (P1 y P7) y en uno de padres consanguíneos (P10). Los tres pertenecían a familias aparentemente no relacionadas, originarias del departamento de Antioquia. En dichos pacientes la ausencia de estos exones se determinó mediante PCR, y fue confirmada en el paciente P1 mediante hibridación genómica comparativa (Comparative Genomic Hybridization, CGH), una técnica ampliamente utilizada para determinar deleciones o inserciones grandes en el genoma37,38.
En este sentido, es importante mencionar que las deleciones y duplicaciones grandes del genAGL se han reportado en pacientes con la enfermedad provenientes de Italia y Egipto25,44; la deleción completa del exón 3 se encontróespecíficamente en dos afroamericanos reportados en el 201026, lo cual indica el potencial deletéreo que tienen este tipo de alteraciones del genoma en la expresión correcta del genAGLy la aparición de esta enfermedad.
El presente estudio constituiría el primer reporte de pacientes con dicha enfermedad que presentan una delecióngrande de los exones 4, 5 y 6. Dado que estos tres pacientes provienen de un área geográfica determinada, en este caso Antioquia, se podría pensar en un efecto fundador; sin embargo, para comprobarlo sería necesario hacer estudios adicionales para determinar el origen de dicha deleción mediante la secuenciación de este mismo locusen el gen AGLa partir de muestras obtenidas de los padres de los pacientes y de otros miembros de las familias. Esto descartaría la presencia de una deleciónde novo y ayudaría a esclarecer el tipo de herencia presente en cada uno de ellos.
Con respecto a los pacientes P6, P8 y P9, en quienes no se evidenciaron mutaciones en los 35 exones y susregiones intrónicas próximas del gen AGL, serían necesarios estudios adicionales que permitan determinar el origen genético del fenotipo clínico que poseen. Por ejemplo, con procedimientos de nueva generación, se podría hacer la secuenciación completa del exoma y el genoma9,45,46 para identificar mutaciones del gen AGLque incluyan los locide regulación de la expresión génica que no se analizaron en este estudio.
Si bien los pacientes analizados presentaban un fenotipo clínico, bioquímico e histopatológico correspondiente a la enfermedad por almacenamiento del glucógeno de tipo III, y su diagnóstico diferencial se basó en la descripción de Kishnani, et al.40, y otros autores, la secuenciación amplia del genoma ayuda a determinar el genotipo mediante la identificación de defectos en distintos genes, por ejemplo, en aquellos relacionados con otros tipos de glucogenosis. Asimismo, se hace patente la necesidad de hacer un diagnóstico molecular completo en aquellos pacientes que poseen enfermedades por almacenamiento de glucógeno.
Es probable que algunas de las mutaciones encontradas en este estudio y ya reportadas anteriormente en el gen AGL, como es el caso de las que inducen la aparición de un condón de parada prematuro, sean procesadas por el mecanismo de degradación del ARN mensajero mediada por secuencias sin sentido (Nonsense Mediated Decay, NMD)47, como se postula en otros estudios en los que no se detectó la presencia de la enzima desramificadora de glucógeno mediante Western blot en muestras de pacientes con la enfermedad por almacenamiento del glucógeno de tipo III con mutaciones similares16.Es importante mencionar que, en el presente trabajo, las mutaciones se clasificaron según su efecto esperado en el procesamiento del ARNm y la secuencia de aminoácidos utilizando los programas computacionales disponibles y que en ninguno de los casos se hicieron análisisin vitro.
Se han reportado varios polimorfismos de un solo nucleótido (SNP) en el gen AGL en la base de datos dbSNP delNational Center for Biotechnology Information (NCBI) y la delExome Aggregation Consortium (ExAC), la mayoría de ellos en regiones intrónicas y en algunos exones6,48,49. En este estudio, se identificaron cinco SNP ya reportadosen otros estudios (rs2307130, rs634880, rs594249, rs2274570 y rs154104), de gran frecuencia alélica en la población.Todos ellos están ubicados en regiones intrónicas y su significación clínica se cataloga como benigna según los predictores bioinformáticos y la dbSNP.
En conclusión, en el presente estudio se analizó el genAGL de diez pacientes colombianos con diagnóstico clínico e histológico de enfermedad por almacenamiento del glucógeno de tipo III, mediante PCR, CGH y secuenciación. Se encontraron mutaciones sin sentido, deleciones de dos nucleótidos y deleciones grandes relacionadas con el fenotipo clínico de los pacientes.
Este es el primer reporte en Colombia, y uno de los primeros a nivel latinoamericano, sobre el espectro de mutaciones del genAGLde pacientes de origen colombiano con esta enfermedad. El estudio permitió confirmar el diagnóstico de la enfermedad, lo cual les garantiza la oportunidad de acceder a tratamientos que mejoren su calidad de vida, además de una correcta asesoría genética para las parejas que están en riesgo de tener hijos con esta enfermedad.
Agradecimientos
Los autores dedican este estudio a la memoria de la doctora Nora Luz Yepes (q.e.p.d.), pediatra gastroenteróloga de la Universidad de Antioquia y profesora del Grupo de Gastrohepatología, quien dedicó su vida a los pacientes y a sus estudiantes de medicina con fervor y total entrega.
Referencias
1. WolfsdorfJI, WeinsteinDA.Glycogen storage diseases. Rev Endocr Metab Disord. 2003;4:95-102.
2. ShinYS.Glycogen storage disease: Clinical, biochemical, and molecular heterogeneity. Semin Pediatr Neurol. 2006;13:115-20. https://doi.org/10.1016/j.spen.2006.06.007
3. OzenH.Glycogen storage diseases: New perspectives. World J Gastroenterol. 2007;13:2541-53. https://doi.org/10.3748/wjg.v13.i18.2541
4. ChengA, ZhangM, OkuboM, OmichiK, SaltielAR.Distinct mutations in the glycogen debranching enzyme foundin glycogen storage disease type III lead to impairment in diverse cellular functions. Hum Mol Genet. 2009;18:2045-52. https://doi.org/10.1093/hmg/ddp128
5. EndoY, FateenE, AoyamaY, HorinishiA, EbaraT, MuraseT, et al.Molecular characterization of Egyptian patients with glycogen storage disease type IIIa. J Hum Genet. 2005;50:538-42. https://doi.org/10.1007/s10038-005-0291-3
6. ShenJJ, ChenYT.Molecular characterization of glycogen storage disease type III. Curr Mol Med. 2002;2:167-75. https://doi.org/10.2174/1566524024605752
7. HaagsmaEB, SmitGP, Niezen-KoningKE, GouwAS, MeermanL, SlooffMJ.Type IIIb glycogen storage disease associated with end-stage cirrhosis and hepatocellular carcinoma. The Liver Transplant Group.Hepatology. 1997; 25:537-40. https://doi.org/10.1002/hep.510250307
8. SentnerCP, HoogeveenIJ, WeinsteinDA, SanterR, MurphyE, McKiernanPJ, et al.Glycogen storage disease type III: Diagnosis, genotype, management, clinical course and outcome. J Inherit Metab Dis. 2016;39:697-704. https://doi.org/10.1007/s10545-016-9932-2
9. Rousseau-NeptonI, OkuboM, GrabsR, ConsortiumFC, MitchellJ, PolychronakosC, et al.A founder AGL mutation causing glycogen storage disease type IIIa in Inuit identified through whole-exome sequencing: A caseseries. CMAJ. 2015;187:E68-73. https://doi.org/10.1503/cmaj.140840
10. MiliA, Ben CharfeddineI, MamaiO, AbdelhakS, AdalaL, AmaraA,et al.Molecular and biochemical characterization of Tunisian patients with glycogen storage disease type III. J Hum Genet. 2012;57:170-5. https://doi.org/10.1038/jhg.2011.122
11. ParvariR, MosesS, ShenJ, HershkovitzE, LernerA, ChenYT.A single-base deletion in the 3’-coding region of glycogen-debranching enzyme is prevalent in glycogen storage disease type IIIA in a population of North African Jewish patients. EurJ Hum Genet. 1997;5:266-70.
12. SanterR, KinnerM, SteuerwaldU, KjaergaardS, SkovbyF, SimonsenH, et al.Molecular genetic basis and prevalence of glycogen storage disease type IIIA in the Faroe Islands. EurJ Hum Genet. 2001;9:388-91. https://doi.org/10.1038/sj.ejhg.5200632
13. TalenteGM, ColemanRA, AlterC, BakerL, BrownBI, CannonRA, et al.Glycogen storage disease in adults. Ann Intern Med. 1994;120:218-26.
14. LucchiariS, SantoroD, PagliaraniS, ComiGP.Clinical, biochemical and genetic features of glycogen debranching enzyme deficiency. Acta Myol. 2007;26:72-4.
15. van HoofF, HersHG.The subgroups of type 3 glycogenosis. Eur J Biochem. 1967;2:265-70. https://doi.org/10.1111/j.1432-1033.1967.tb00134.x
16. DingJH, de BarsyT, BrownBI, ColemanRA, ChenYT.Immunoblot analyses of glycogen debranching enzyme in different subtypes of glycogen storage disease type III. J Pediatr. 1990;116:95-100.
17. Yang-FengTL, ZhengK, YuJ, YangBZ, ChenYT, KaoFT.Assignment of the human glycogen debrancher gene to chromosome 1p21. Genomics. 1992;13:931-4. https://doi.org/10.1016/0888-7543(92)90003-B
18. YangBZ, DingJH, EnghildJJ, BaoY, ChenYT.Molecular cloning and nucleotide sequence of cDNA encoding human muscle glycogen debranching enzyme. J Biol Chem. 1992;267:9294-9.
19. BaoY, YangBZ, DawsonTLJr, ChenYT.Isolation and nucleotide sequence of human liver glycogen debranching enzyme mRNA: Identification of multiple tissue-specific isoforms. Gene. 1997;197:389-98. https://doi.org/10.1016/S0378-1119(97)00291-6
20. LucchiariS, FoghI, PrelleA, PariniR, BresolinN, MelisD, et al.Clinical and genetic variability of glycogen storage disease type IIIa: Seven novel AGL gene mutations in the Mediterranean area. Am J Med Genet. 2002;109:183-90. https://doi.org/10.1002/ajmg.10347
21. HorinishiA, OkuboM, TangNL, HuiJ, ToKF, MabuchiT, et al.Mutational and haplotype analysis of AGL in patients with glycogen storage disease type III. J Hum Genet. 2002;47:55-9. https://doi.org/10.1007/s100380200000
22. OkuboM, HorinishiA, SuzukiY, MuraseT, HayasakaK.Compound heterozygous patient with glycogen storage disease type III: Identification of two novel AGL mutations, a donor splice site mutation of Chinese origin and a 1-bp deletion of Japanese origin. Am J Med Genet. 2000;93:211-4. https://doi.org/10.1016/S0378-1119(97)00291-6
23. OkuboM, HorinishiA, TakeuchiM, SuzukiY, SakuraN, HasegawaY, et al.Heterogeneous mutations in the glycogen-debranching enzyme gene are responsible for glycogen storage disease type IIIa in Japan. Hum Genet. 2000;106:108-15.
24. OgimotoA, OkuboM, OkayamaH, ShinYS, EndoY, EbaraT, et al.A Japanese patient with cardiomyopathy caused by a novel mutation R285X in the AGLgene. Circ J. 2007;71:1653-6. https://doi.org/10.1253/circj.71.1653
25. EndoY, FateenE, El ShabrawyM, AoyamaY, EbaraT, MuraseT, et al.Egyptian glycogen storage disease type III - identification of six novel AGL mutations, including a large 1.5 kb deletion and a missense mutation p.L620P with subtype IIId. Clin Chem Lab Med. 2009;47:1233-8. https://doi.org/10.1515/CCLM.2009.281
26. GoldsteinJL, AustinSL, BoyetteK, KanalyA, VeerapandiyanA, RehderC, et al.Molecular analysis of the AGL gene: Identification of 25 novel mutations and evidence of genetic heterogeneity in patients with glycogen storage disease type III. Genet Med.2010;12:424-30. https://doi.org/10.1097/GIM.0b013e3181d94eaa
27. AoyamaY, OzerI, DemirkolM, EbaraT, MuraseT, PodskarbiT, et al.Molecular features of 23 patients with glycogen storage disease type III in Turkey: A novel mutation p.R1147G associated with isolated glucosidase deficiency, along with 9 AGLmutations. J Hum Genet. 2009;54:681-6. https://doi.org/10.1038/jhg.2009.100
28. SentnerCP, VosYJ, Niezen-KoningKN, MolB, SmitGP.Mutation analysis in glycogen storage disease type III patients in the Netherlands: Novel genotype-phenotype relationships and five novel mutations in the AGL gene. JIMD Rep. 2013;7:19-26. https://doi.org/10.1007/8904_2012_134
29. ZimmermannA, RossmannH, BucerzanS, Grigorescu-SidoP.A novel nonsense mutation of the AGL gene in a Romanian patient with glycogen storage disease type IIIa. Case Rep Genet. 2016;2016:8154910. https://doi.org/10.1155/2016/8154910
30. RhoumaFB, MessaiH, HsounaS, HalimNB, CherifW, FadhelSB, et al.History of settlement of villages from Central Tunisia by studying families sharing a common founder glycogenosis type III mutation. Mitochondrial DNA A DNA MappSeq Anal. 2016;27:3194-8. https://doi.org/10.3109/19401736.2015.1007331
31. LuC, QiuZ, SunM, WangW, WeiM, ZhangX.Spectrum of AGL mutations in Chinese patients with glycogen storage disease type III: Identification of 31 novel mutations. J Hum Genet. 2016;61:641-5. https://doi.org/10.1038/jhg.2016.24
32. KumarP, HenikoffS, NgPC.Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073-81. https://doi.org/10.1038/nprot.2009.86
33. AdzhubeiI, JordanDM, SunyaevSR.Predicting functional effect of human missense mutations using PolyPhen-2.Curr Protoc Hum Genet. 2013;7:20https://doi.org/10.1002/ 0471142905.hg0720s76
34. KircherM, WittenDM, JainP, O’RoakBJ, CooperGM, ShendureJ.A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310-5. https://doi.org/10.1038/ng.2892
35. den DunnenJT, DalgleishR, MaglottDR, HartRK, GreenblattMS, McGowan-JordanJ, et al.HGVS Recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564-9. https://doi.org/10.1002/humu.22981
36. den DunnenJT.Sequence variant descriptions: HGVS nomenclature and mutalyzer. Curr Protoc Hum Genet. 2016;90:7.13.1-7.13.19. https://doi.org/10.1002/cphg.2
37. AradhyaS, LewisR, BonagaT, NwokekehN, StaffordA, BoggsB, et al.Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet Med.2012;14:594-603. https://doi.org/10.1038/gim.2011.65
38. RettererK, ScuffinsJ, SchmidtD, LewisR, Pineda-ÁlvarezD, StaffordA, et al.Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med.2015;17:623-9. https://doi.org/10.1038/gim.2014.160
39. DagliA, SentnerCP, WeinsteinDA. Glycogen storage disease type III. In: PagonRA, AdamMP, ArdingerHH, WallaceSE, AmemiyaA, BeanLJ, et al., editors. Gene Reviews. Washington, D.C.: University of Washington; 1993.
40. KishnaniPS, AustinSL, ArnP, BaliDS, BoneyA, CaseLE, et al.Glycogen storage disease type III diagnosis and management guidelines. Genet Med.2010;12:446-63. https://doi.org/10.1097/GIM.0b013e3181e655b6
41. ColemanRA, WinterHS, WolfB, GilchristJM, ChenYT.Glycogen storage disease type III (glycogen debranching enzyme deficiency): Correlation of biochemical defects with myopathy and cardiomyopathy. Ann Intern Med. 1992;116:896-900. https://doi.org/10.7326/0003-4819-116-11-896
42. MaireI, BaussanC, MoattiN, MathieuM, LemonnierA.Biochemical diagnosis of hepatic glycogen storage diseases: 20 years Frenchexperience. Clin Biochem. 1991;24:169-78. https://doi.org/10.1016/0009-9120(91)90511-C
43. LucchiariS, PagliaraniS, SalaniS, FilocamoM, Di RoccoM, MelisD, et al.Hepatic and neuromuscular forms of glycogenosis type III: Nine mutations in AGL. Hum Mutat. 2006;27:600-1. https://doi.org/10.1002/humu.9426
44. LucchiariS, DonatiMA, MelisD, FilocamoM, PariniR, BresolinN, et al.Mutational analysis of the AGL gene: Five novel mutations in GSD III patients. Hum Mutat. 2003;22:337.https://doi.org/10.1002/humu.9177
45. ZhangW, CuiH, WongLJ.Application of next generation sequencing to molecular diagnosis of inherited diseases. Top Curr Chem. 2014;336:19-45. https://doi.org/10.1007/128_2012_325
46. YuberoD, BrandiN, OrmazabalA, García-CazorlaA, Pérez-DueñasB, CampistolJ, et al.Targeted next generation sequencing in patients with inborn errors of metabolism. PLoS One. 2016;11:e0156359. https://doi.org/10.1371/journal.pone.0156359
47. SilvaAL, RomaoL.The mammalian nonsense-mediated mRNA decay pathway: To decay or not to decay! Which players make the decision?FEBS Lett. 2009;583:499-505. https://doi.org/10.1016/j.febslet.2008.12.058
48. HorinishiA, MuraseT, OkuboM.Novel intronic polymorphisms (IVS6-73A/G and IVS21+124A/G) in the glycogen-debranching enzyme (AGL) gene. Hum Mutat. 2000;16:279. https://doi.org/10.1002/1098-1004(200009)16:3<279::AID-HUMU32>3.0.CO;2-X
49. ShamseldinHE, Al-DosariM, Al-JbaliL, RahbeeniZ, QariA, HashemM, et al.Study of consanguineous populations can improve the annotation of SNP databases. Eur J Med Genet. 2011;54:118-20.https://doi.org/10.1016/j.ejmg.2010.10.009
Apéndice
Características clínicas y moleculares más importantes de los subtipos de la enfermedad por almacenamiento de glucógeno de tipo III (EDG III)
Apéndice
Protocolos de PCR para amplificar los exones del gen AGL
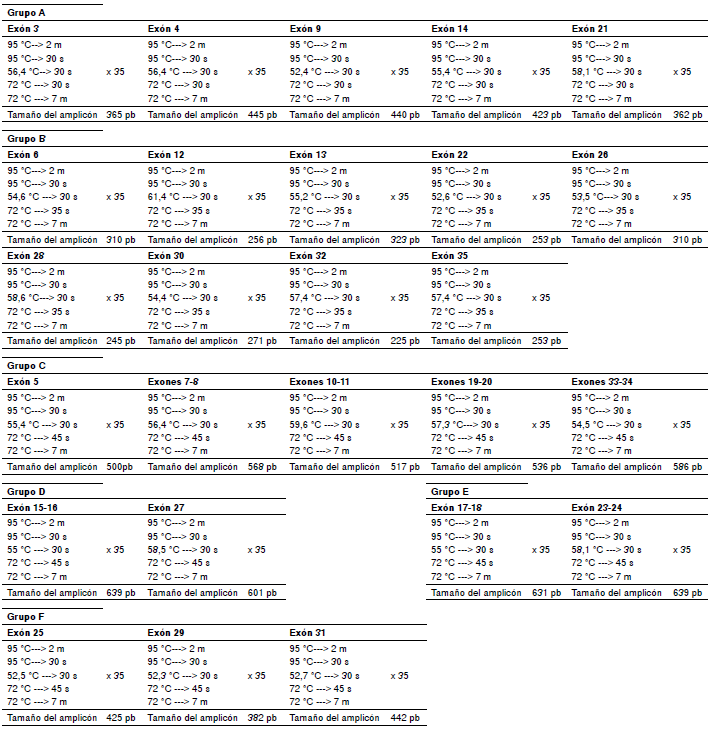
Apéndice
Oligonucleótidos utilizados para amplificar los exones del gen AGL mediante PCR, con base en el artículode Goldstein, et al., 2010

Notas
Notas de autor
*Correspondencia: Andrés Augusto Arias, Grupo de Inmunodeficiencias Primarias,Sede de Investigación Universitaria, Universidad de Antioquia,Calle 70 N° 52-21, oficina 530, Medellín, ColombiaTeléfono: (574) 219 6471 aaugusto.arias@udea.edu.co
Declaración de intereses
