Resumen: En este estudio, usando la Teoría del Funcional de la Densidad (DFT), las cargas NBO y la aproximación de la supermolécula, se han podido determinar las interacciones moleculares entre el polietileno lineal de baja densidad (PELBD) y el quitosano (Q), usando la modificación química del PELBD con acrilamida (AAm) como agente compatibilizante (PELBD-g-AAm). Los resultados obtenidos indican que en las moléculas de la mezcla de PELBD/Q están actuando fuerzas intermoleculares muy débiles del tipo Debye, mientras que entre las moléculas de Q/PELBD-g-AAm se puede formar un complejo "estable" debido a la presencia de enlaces de hidrógeno.
Palabras clave:cargas NBOcargas NBO, DFT DFT, interacciones moleculares interacciones moleculares, supermolécula supermolécula, quitosano quitosano.
Abstract: By using the Density Functional Theory (DFT), NBO charges and the “supermolecule”approach we have been able to determine the molecular interactions between low density linear polyethylene (LLDPE) and chitosan (Q), employing the chemical modification of PELBD with acrylamide (AAm) as a compatibilizing agent LLDPE-g-AAm). The obtained results indicate that in the molecules of the LLDPE/Ch blend are acting very weak intermolecular forces of Debye type, while among molecules of Q/ LLDPE-g-AAm blend may be forming a "stable" complex due to the presence of hydrogen bonds.
Keywords: NBO charges, DFT, Molecular Interacctions, Supermolecule, Chitosan.
Artículos Científicos
Interacciones moleculares entre matrices poliméricas de PELBD/Quitosano/PELBD-g-Acrilamida: Estudio computacional*
Molecular interactions between polymer matrices of LLDPE/Chitosan/PELBD-g-Acrylamide: Computational study
Universidad de los Andes

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 3.0 Internacional.
Recepción: 23 Noviembre 2017
Revisado: 30 Noviembre 2017
Aprobación: 28 Diciembre 2017
Las interacciones moleculares son las responsables de las propiedades únicas de sustancias tan simples como el agua, hasta las más complejas como el ADN, macromoléculas sintéticas, entre otras. En el caso de polímeros, las interacciones moleculares juegan un papel crucial tanto en sus propiedades mecánicas como en su uso para procesos de interés, de aquí el desarrollo teórico de distintas técnicas para calcular los parámetros de interacción.
Hoy en día, existe un marcado interés por el estudio y la síntesis de materiales constituidos por polímeros biodegradables a partir de fuentes naturales como el quitosano1, un polisacárido natural que, consiste de unidades 2-acetamido-2-desoxi-β-D-glucosa y 2-amino-2-desoxi-β-D-glucosa unidas por enlaces tipo β(1→ 4). Las propiedades catiónicas del quitosano ofrecen buenas oportunidades para aprovechar la interacción que pueden establecer sus electrones con numerosos compuestos, siendo uno de los biomateriales más versátiles de la naturaleza2,3. Sin embargo, generalmente las mezclas entre poliolefinas como el polietileno y biopolímeros como el Q presentan incompatibilidad, por lo que para favorecer las interacciones de las mezclas usualmente se incorpora un agente compatibilizante4,5.
La compatibilidad, biocompatibilidad, y los parámetros de mezclas están intrínsecamente relacionados con el grado de interacción entre los componentes de la mezcla y éstos, a su vez, con las interacciones moleculares, de aquí que el éxito en § la modelación de procesos industriales dependa, en forma decisiva, de una descripción eficaz de las propiedades termodinámicas y del comportamiento de fase de los sistemas químicos asociados. La química computacional puede ayudar a predecir los tipos de interacciones moleculares, a través de cálculos mecano-cuánticos, contribuyendo a la racionalización de la síntesis y la predicción de sus propiedades6,7, lo que permite sentar las bases y abaratar los costos de futuras investigaciones.
En trabajos anteriores, se estudiaron los enlaces de hidrógeno del fenol en solución8, debido a que es capaz de formar este tipo de enlaces fácilmente, lo que lo convierte en un modelo adecuado para estudiar estas interacciones mediante la química computacional a nivel de teoría DFT. En particular se realizó, la optimización de su geometría y cálculos de frecuencias vibracionales, para asignar los picos observados en el espectro IR, en el cual se evidenció que se desplazó la banda correspondiente al estiramiento del grupo hidroxilo OH a un número de onda más bajo, debido a que con el aumento de la concentración del fenol, se favorece la formación de enlaces de hidrógeno intermoleculares. Estos resultados fueron corroborados por la predicción de las frecuencias vibracionales obtenidas a través de datos computacionales, demostrándose así la utilidad de las derivaciones que predice la química computacional en el estudio de las interacciones.
Así mismo, también se ha reportado en una investigación reciente, el estudio de una mezcla de quitosano y un polietileno funcionalizado, por medio de la combinación de los resultados experimentales observados en el espectro infrarrojo y los cálculos de frecuencias vibracionales empleando el modelaje molecular9. Los autores pudieron asignar las señales en el IR y detectar las interacciones que influyen en la frecuencia vibratoria de los grupos funcionales de la mezcla.
En este trabajo, se usará la Teoría del Funcional de la Densidad (DFT), las cargas NBO y la aproximación de la super-molécula, para determinar las interacciones moleculares entre el PELBD y el Q, usando la modificación química del PELBD con acrilamida, como agente compatibilizante (PELBD-g-AAm). Hoy en día, este tipo de métodos “in silico” permite el estudio de las propiedades moleculares y electrónicas de polímeros10,11,12,13, abriendo nuevas perspectivas para el estudio de materiales poliméricos biocompatibles.
Las estructuras modelo optimizadas para el uso de los métodos computacionales, fueron: (a) un olígomero de PELBD, (b) un dímero de quitosano, y (c) un agente compatibilizante constituido por etileno enlazado a una unidad de AAm (figura 1). Aunque se arrastran diferentes errores en estas aproximaciones como la presencia de los dos grupos CH3 en los extremos del quitosano, el principal objetivo del estudio fue evaluar si la presencia del agente compatibilizante favorecería nuevas interacciones de tipo enlace de hidrógeno entre las unidades del PELBD y las del quitosano, para lo cual estos modelos son útiles, como han demostrado otros autores previamente14,15. Por otra parte, se ha demostrado que, para moléculas grandes como quitosano, el modelaje molecular usando, por lo menos, un dímero, reproduce los resultados experimentales de manera aceptable, fundamentalmente, las bandas armónicas de vibración15,16,17,18.
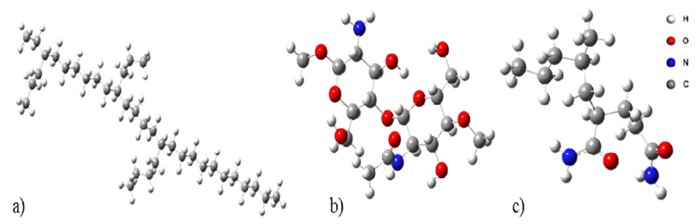
Fig. 1
Estructuras optimizadas de a) PELBD, b) quitosano y c) PELBD-g-AAm (agente compatibiilizante)
El cálculo de las estructuras electrónicas de mínima energía para el PELBD, PELBD-g-AAm, Q, PELBD/Q y Q/PELBDg-AAm fue llevado a cabo utilizando la Teoría del Funcional de la Densidad (DFT), usando como conjunto funcional-base en el presente estudio: B3LYP/6-31++G(d,p), incluido en el paquete computacional Gaussian 09 para Windows.
El criterio de convergencia utilizado en la matriz densidad, fue de 10-9 unidades atómicas, el valor para el desplazamiento máximo fue de 0,0018 Å, y para la fuerza máxima de 0,00045 Hartree/Bohr. Los puntos estacionarios de mínima energía y los correspondientes al estado de transición, fueron verificados usando cálculos de frecuencia, la ausencia de frecuencias negativas garantiza un mínimo en la superficie de energía potencial10.
Las magnitudes termodinámicas, como la energía de punto cero (ZPVE) y entropías absolutas, S(T), fueron obtenidas a partir de cálculos de frecuencias (armónicas) haciendo uso de la mecánica cuántica y la mecánica estadística para obtener las funciones de partición de las especies10.
Para el cálculo de la energía de interacción en las mezclas PELBD/Q y PELBD-g-AAm/Q, se usó la teoría de la supermolécula, utilizando la corrección de contraposición11. Para ello, se empleó la siguiente ecuación:
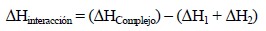 (1)
(1)donde ΔHComplejo= entalpía de la supermolécula formada por las especies interactuantes, ΔH1 = entalpia de formación de la molécula 1 y ΔH2 = entalpia de formación de la molécula 2. Para corregir estos valores, se usó la corrección de contraposición con el propósito de tener valores reales de la interacción11.
Un valor muy positivo de ΔHinteracción significará que existe “dispersión” en las moléculas y, por lo tanto, no existirá probabilidad de que se forme un complejo “estable”. Por el contrario, valores negativos indicarán estabilización por interacción dipolo-dipolo11.
Las cargas NBO fueron calculadas con el propósito de analizar las superficies polares y no polares de las moléculas interactuantes. Para esto, se hizo uso del programa NBO 4.0 instalado dentro del paquete computacional Gaussian 09W.
La figura 2 muestra las geometrías optimizadas de las moléculas PELBD/Q y Q/PELBD-g-AAm, sus estructuras moleculares y la numeración de sus átomos.
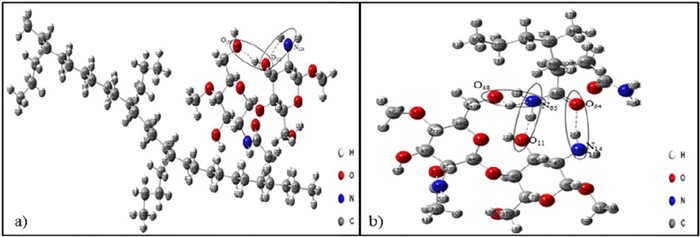
Fig. 2
Estructura optimizada del a) PELBD/Q y b) Q/PELBD-g-AAm, mediante el método B3LYP/6-31G++ (d,p)
En la estructura optimizada de la mezcla PELBD/Q se observa en el quitosano la presencia de enlaces de hidrógeno intramoleculares. Tomando en cuenta los radios de Van der Waals se considera la presencia de enlace de hidrógeno entre O121-H122···O158 cuya distancia es de 1,9455 Å, en donde el hidrógeno H122 está interaccionando con el O158. Sin embargo, la distancia del enlace N124-H126···O121 indica la presencia de un enlace más débil (2,3519 Å). Por otra parte, el ángulo N124-H126-O121 es de 105,58°. Estas interacciones son propiciadas por los grupos hidroxilo y amida adyacentes en la molécula.
Al evaluar las estructuras optimizadas de Q/PELBD-g-AAm se observa que el agente compatibilizante, proporciona segmentos capaces de promover interacciones intermoleculares con el quitosano a través de enlaces de hidrógeno. Tomando en cuenta los radios de Van der Waals, se considera la presencia de enlaces de hidrógeno entre los grupos funcionales amino (NH2) y carbonilo (C=O) del PELBD-g-AAm con las aminas (NH2) e hidroxilos (OH) del quitosano.
Observando los grupos NH2 de la amida y los OH del quitosano se aprecia que la distancia entre O48-H48···N65 es de 2,3400 Å donde el hidrógeno H48 está interaccionando con el N65. Las distancias entre estos átomos favorecen una interacción del tipo enlace de hidrógeno. Finalmente el grupo carbonilo (C=O) de la amida, puede estar interactuando con un grupo amino del quitosano, debido a que la distancia entre O64···H16-N14 es de 2,0100 Å, además el ángulo O64-H16-N14 es de 8,91°, lo cual también puede promover la formación de un enlace de hidrógeno intermolecular.
Para el cálculo de la energía de interacción entre PELBD/Q y Q/PELBD-g-AAm se usó la teoría de la supermolécula, usando la corrección de contraposición9, para ello, se usó la ecuación 1. Los valores obtenidos se resumen en la tabla 1.
Propiedades termodinámicas de los polímeros a 298,15 K
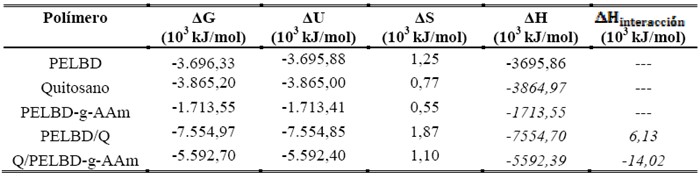
El valor positivo del ΔHinteracción obtenido por el método de la supermolécula indica que en las moléculas PELBD/Q actúan fuerzas de Debye, siendo estas fuerzas intermoleculares muy débiles, por lo que no se puede formar un complejo “estable.
Para entender la interacción dipolo-dipolo inducido en la mezcla, hay que recordar la naturaleza mecanocuántica de los orbitales atómicos y moleculares, donde un orbital es una zona alrededor del átomo que encierra una determinada probabilidad de encontrar a los electrones12. A tiempos sufícientemente cortos, se puede considerar que la nube electrónica fluctúa creando momentos dipolares instantáneos16. Por lo tanto, una molécula polar como el quitosano, cuyo momento dipolar calculado en este trabajo = 4,21 D resultó similar al calculado para un dímero en un trabajo previo19, puede inducir un dipolo en otra molécula apolar como el PELBD (0,17 D, valor calculado en este trabajo). Sin embargo, la energía de interacción será comparativamente menor dado que la carga presente también lo es.
En el caso de las moléculas Q/PELBD-g-AAm el valor negativo del ΔHinteracción obtenido por el método de la super-molécula indica que puede estarse formando un complejo “estable”, debido a la presencia del agente compatibilizante. Al analizar la interacción entre las moléculas se puede inferir que se produce la formación de enlaces de hidrógeno entre los grupos funcionales del quitosano y la amida injertada en el PELBD.
Con el propósito de analizar las superficies polares y no polares de las moléculas interactuantes, se calcularon las cargas NBO. Al evaluar la unidad repetitiva (CH2-CH2)n de la molécula de PELBD, se encuentra como resultado una molécula apolar. Por otra parte, el análisis de las cargas de los grupos funcionales de la molécula de quitosano confirma su polaridad y la presencia de enlaces de hidrógeno.
Al analizar las cargas de la molécula de quitosano es necesario tomar en cuenta diversos factores. Así pues, se evidencia que el átomo N124 (figura 3)-correspondiente a la amina, posee la carga más elevada de la molécula, debido a que tiene localizados un par de electrones no enlazantes. En el caso del átomo N144 (figura 3) de la amida la carga será menor, esto es causado porque que hay una estructura contribuyente donde el par de electrones no compartido del átomo de nitrógeno se deslocaliza hacia el carbonilo.
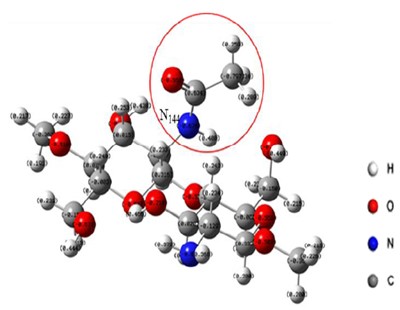
Fig. 3
Estructura optimizada de la amida del Q mediante el método B3LYP/6-31G++ (d,p)
En el oxígeno O133 perteneciente al enlace glucosídico del quitosano se observó una menor densidad de carga, posiblemente por un efecto estéreo-electrónico, el cual se interpreta como una interacción del par electrónico del oxígeno glucosídico, con el extremo del orbital antienlazante del enlace σC1-O5 del anillo piranósico de la unidad monomérica.
Los enlaces O-H están muy polarizados por la electronegatividad del oxígeno y por el hecho de que el único protón del núcleo del hidrógeno atrae débilmente a los electrones del enlace; los grupos OH actúan como donadores de hidrógeno lo que propicia la formación de enlaces de hidrógeno entre los grupos funcionales adyacentes, por lo que la densidad de carga de los átomos O152 y O158 tiende a ser menor que la del nitrógeno de la amina (N124), el cual es mejor aceptor.
Se observa que el enlace O121δ- - H122δ+ es uno de los más polarizados de la molécula, promoviendo la formación de este tipo de enlaces con el O158 (O121δ--H122δ+···O158δ-). En el caso del átomo de nitrógeno a pesar de ser más aceptor, también puede favorecerse el enlace hidrógeno entre O121δ-•••H126δ+ - N121δ-. Estos valores se muestran en las tablas 2, 3 y 4.
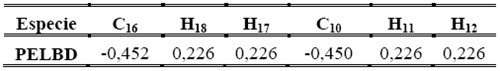
Cargas NBO principales del PELBD, empleando el método/base B3LYP/6-31G++ (d,p)
Cargas NBO principales del Q, empleando el método/base B3LYP/6-31G++ (d,p)

Cargas NBO principales de una mezcla PELBD/Q, empleando el método/base B3LYP/6-31G++ (d,p)

Las cargas NBO de las especies que interaccionan en la mezcla PELBD/Q, confirman las fuerzas de Debye, las cuales se favorecen por la polaridad y apolaridad de las moléculas de quitosano y polietileno, respectivamente.
Al estudiar las cargas del grupo amida del agente compatibilizante, se observa un efecto peculiar reportado en trabajos previos13, donde la densidad electrónica del N65 es mayor con respecto a la del O64,, esto se debe a un fenómeno llamado “back transfer” o retro-transferencia, el cual es favorecido cuando la amida se encuentra en una conformación planar. Esto surge, en parte, debido a que la electronegatividad del nitrógeno atrae electrones a lo largo del enlace σ de C-N, en la dirección opuesta al desplazamiento que ocurre cuando el par solitario del nitrógeno es compartida con el átomo de carbono del carbonilo a través del enlace π.
En la amida injertada en el PELBD se evidencia que el átomo de nitrógeno (N65) posiblemente esté actuando como el principal aceptor de protones, lo cual favorece la formación de enlaces de hidrógeno con los hidroxilos O48 δ-H49 δ+(O48 δ-H49 δ+···N65 δ-) (figura 2.b). Por su parte, el O48 tiene una densidad electrónica menor a la del resto, lo que confirma la formación de este tipo de enlace. En la figura 2.b también se observa que, en el caso del átomo de oxígeno (O64) del carbonilo de la acrilamida, la situación es similar excepto por la menor densidad de carga; sin embargo, también se favorece el enlace entre O64 δ-···H16 δ+-N14 δ-ya que los orbitales del oxígeno tienen una densidad de carga negativa alta, y por consiguiente se pueden unir a la carga positiva del hidrógeno.
Finalmente, las cargas NBO de las moléculas Q/PELBD-g-AAm confirman los resultados obtenidos, los cuales permiten proponer los enlaces de hidrógeno que pueden favorecerse entre el quitosano y la acrilamida. El resumen de estos valores se muestra en las tablas 5 y 6.
Cargas NBO principales del PELBD-g-AAm, empleando el método/base B3LYP/6-31G++ (d,p)

Cargas NBO principales de las moléculas Q/PELBD-g-AAm, empleando el método/base B3LYP/6-31G++ (d,p)

-
El modelaje molecular realizado a nivel de teoría DFT/B3LYP/6-31++G (d,p), de las moléculas PELBD/Q y Q/PELBD-g-AAm, permitió determinar a través del método de la supermolécula que puede estarse formando un complejo “estable” en el caso de las moléculas Q/PELBD-g-AAm, debido a las interacciones entre el quitosano y la acrilamida. En el caso de las moléculas PELBD/Q se determinó que en estas actúan fuerzas de Debye, por lo que no se puede formar un complejo “estable”.
-
Mediante la optimización geométrica se pudieron proponer diversas interacciones del tipo enlace de hidrógeno entre la amida del agente compatibilizante y los grupos OH y NH2 del quitosano.
-
El análisis de las cargas NBO de las especies interactuantes confirmaron los resultados obtenidos por el método de la súpermolécula.
-
Finalmente, en esta investigación se concluye que la química computacional puede ayudar a proponer los tipos de interacciones moleculares, a través de cálculos mecanocuánticos, contribuyendo a la racionalización de la síntesis y a la predicción de sus propiedades, lo que permite sentar las bases y abaratar los costos de futuras investigaciones.
Se agradece al FONACIT por el apoyo financiero a través del proyecto F-2013000395, a la UDO por el Proyecto CI-2-0404001673/10 y por el Proyecto POA-AC020120783.
Fig. 1
Estructuras optimizadas de a) PELBD, b) quitosano y c) PELBD-g-AAm (agente compatibiilizante)
Fig. 2
Estructura optimizada del a) PELBD/Q y b) Q/PELBD-g-AAm, mediante el método B3LYP/6-31G++ (d,p)
Propiedades termodinámicas de los polímeros a 298,15 K
Fig. 3
Estructura optimizada de la amida del Q mediante el método B3LYP/6-31G++ (d,p)
Cargas NBO principales del PELBD, empleando el método/base B3LYP/6-31G++ (d,p)
Cargas NBO principales del Q, empleando el método/base B3LYP/6-31G++ (d,p)
Cargas NBO principales de una mezcla PELBD/Q, empleando el método/base B3LYP/6-31G++ (d,p)
Cargas NBO principales del PELBD-g-AAm, empleando el método/base B3LYP/6-31G++ (d,p)
Cargas NBO principales de las moléculas Q/PELBD-g-AAm, empleando el método/base B3LYP/6-31G++ (d,p)
