Nota Tecnica
Descarboxilación de dos ésteres alílicos de ácidos areno carboxílicos mediada por catalizadores de paladio, hacia la síntesis de 3,4-dialcoxitiofenos
Descarboxilación de dos ésteres alílicos de ácidos areno carboxílicos mediada por catalizadores de paladio, hacia la síntesis de 3,4-dialcoxitiofenos
Avances en Química, vol. 13, núm. 2, pp. 33-40, 2018
Universidad de los Andes

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 3.0 Internacional.
Recepción: 21 Mayo 2018
Revisado: 05 Julio 2018
Aprobación: 06 Agosto 2018
Resumen: Se sintetizaron dos moléculas modelo el dimetoxibenzoato de alilo (1) y el 2,5-dicarbaliloxi-3,4-etilendioxitiofeno (2a). Estas se sometieron a las condiciones de reacción de descarboxilación mediada por Pd0 para β-cetoéteres alílicos. La eliminación de CO2 del compuesto 1 solo fue posible cuando un segundo catalizador de PdII se adicionó a la reacción. Bajo estas condiciones fue posible la formación de un producto de homoacoplamiento con impedimento estérico 3. Una especie electrofílica de PdII parece ser necesaria para generar productos de descarboxilación aromática desde un éster alílico. Sin embargo, la reacción requiere calentamiento prolongado y no alcanza un alto rendimiento.
Palabras clave: descarboxilación, areno, alcoxitiofeno, catalizador de Pd, éster alílico, biaril.
Abstract: Two model molecules, allyl 2,6-dimethoxybenzoate (1) and 2,5-dicarballyloxy-3,4-ethylendioxy-thiophene (2a), were synthesized. These were subjected to the same conditions as the decarboxylation of allyl-β-ketoesters mediated by Pd0 catalyst. The CO2 elimination for 1 was possible only when a second catalyst of PdII was added. Under these conditions a homocoupling product (3) was possible. An electrophilic PdII species seems to be necessary to generate the decarboxylated products, but this requires a long reaction time and the yields are moderated.
Keywords: Decarboxylation, Arene, Aalkoxythiophene, Pd catalyst, Allyl ester, Biaryl.
Introducción
La descarboxilación aromática es una reacción catalizada por metales ampliamente usada en la síntesis de compuestos aromáticos1. Existen tres principales metodologías de descarboxilación basadas en catalizadores de cobre2-5, plata6-7 y Pd8. La última aplicada solamente en ácidos de arenos con grupos electrodonadores. Los ácidos carboxílicos de arenos sustituidos son una alternativa a los compuestos organometálicos en la síntesis de compuestos biarílicos, por medio de reacciones de acoplamiento descarboxilativo9-10. La descarboxilación con el sistema catalítico Ag2CO3/AcOH ha sido utilizada con éxito en la preparación de sistemas 3,4-dialcoxitiofeno11. Estos compuestos son útiles en la fabricación de materiales electrónicos orgánicos12-17. Sin embargo, con esta metodología no se puede obtener el producto de doble descarboxilación del ácido 2-feniltieno[3,4-d][1,3]dioxol-4,6-dicarboxílico. Este diácido sufre una transformación espontánea a temperaturas más altas que 50 °C en el disolvente DMSO11. La reacción de descarboxilación para este diácido catalizada por Pd(CF3COO)2, con DMSO como ligante y DMF como disolvente 70 °C, tampoco ha dado resultado18. Por lo tanto, una metodología de descarboxilación que ocurra a una temperatura menor que 50 °C y/o que sea compatible para grupos sensibles al medio básico, es necesaria.
Por otro lado, la reacción de descarboxilación mediada por Pd0 de los β-cetoésteres alílicos ocurre a temperatura ambiente y condiciones suaves19. Estas condiciones de reacción han sido aplicadas en la reacción de alilación descarboxilativa de ésteres alílicos de ácidos acetilénicos (obteniéndose productos con enlace C-C sp-sp3)20 y de ácidos de cumarinas (obteniéndose productos con enlace C-C sp2-sp3)21. Estos ejemplos indican que la reacción de descarboxilación de ésteres alílicos puede ser llevada a cabo sin la presencia de un grupo ceto en la posición β del correspondiente grupo carboxilo.
Se puede visualizar una posible ventaja de la reacción de descarboxilación de ésteres alílicos en la preparación de 3,4-dialcoxitiofenos (esquema 1). Esta consiste en eliminar los pasos de reacción saponificación y acidificación de la ruta de síntesis típica22-23, haciendo que la obtención de estos compuestos sulfurados sea más directa. Además, se podrían instalar en la etapa de O-alquilación sustituyentes tipo 5 que tengan grupos funcionales sensibles al medio básico o ácido. En este trabajo, se informan los resultados alcanzados en la descarboxilación mediada por Pd de dos ésteres alílicos de areno y tiofeno.
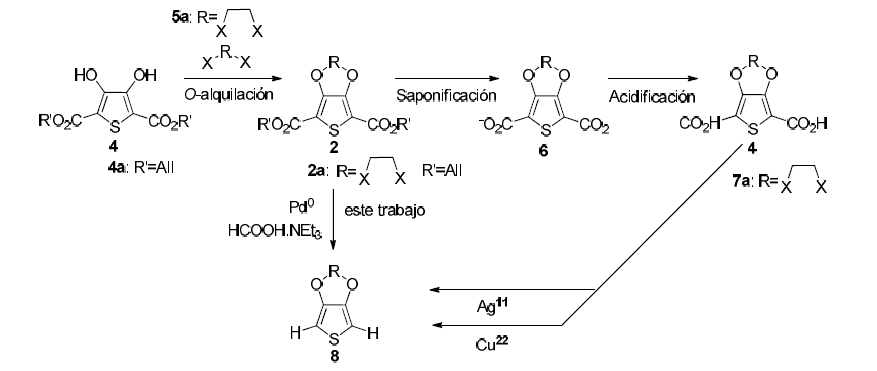
Esquema 1
Preparación de 3,4-dialcoxitiofenos a través de una reacción descarboxilación aromática mediada por metales. El protocolo de descarboxilación clásico se hace via los compuestos 2-6-7; en este trabajo se propone hacerlo mediante la reacción de descarboxilación de ésteres dialílicos 2a
Parte experimental
Información general
Los espectros de 1H RMN fueron obtenidos a 300 MHz en un espectrómetro Bruker Avance, a 500 MHz en un espectrómetro Varian NMRSystem y fueron referenciados al pico del disolvente no deuterado residual de 7,26 ppm (CDCl3). Los espectros de 13C RMN, fueron obtenidos a 75 MHz en un espectrómetro Bruker Avance, a 125 MHz en un espectrómetro Varian NMR System y fueron referenciados respecto al pico del disolvente residual en 77 ppm (CDCl3).
Síntesis de tiodiglicolato de dialilo 14
En un matraz de fondo redondo de 3 bocas de 250 mL se colocaron 2,30 g (15 mmol) de ácido tiodiglicólico 98 %, 5 mL (4,27 g, 72,4 mmol) de alcohol alílico y 0,171 g (0,9 mmol) de ácido p-toluenfsulfónico monohidratado y 80 mL de benceno. A continuación, se colocó una trampa Dean Stark y se calentó en baño de aceite a temperatura de reflujo. El calentamiento se mantuvo hasta recolectar 1 mL de agua en el fondo de la trampa, esto duró 3 h. La mezcla se enfrío a temperatura ambiente y se lavó con 2 fracciones de 25 mL de una solución saturada de NaHCO3 y después con 25 mL de salmuera. Se secó con Na2SO4 anhidro y se evaporó el disolvente a presión reducida, obteniéndose 2,83 g (12,3 mmol, 82%) de un líquido amarillo. 1H RMN (500 MHz, CDCl3) δ (ppm): 5,97-5,89 (ddd, 2H, J3= 6 Hz), 5,37-5,34 (d, 2H, J1= 17 Hz), 5,28-5,26 (d, 2H, J2= 10,5 Hz) 4,65-4,63 (d, 4H, J= 6 Hz), 3,42 (s, 4H). 13C RMN (125 MHz, CDCl3) δ (ppm): 169,4; 131,5;118,8; 66,0; 33,3. IR ν (cm-1): 2945, 1729, 1273, 1120, 986, 927, 682. EM-IE, [M]= 230; C10H14O4S: 230,06.
Síntesis de oxalato de dialilo 15
En un matraz de fondo redondo de 3 bocas de 250 mL se colocaron 1,89 g (15 mmol) de ácido oxálico dihidratado, 5 mL (4,27 g, 72,4 mmol) de alcohol alílico y 0,171 g (0,9 mmol) de ácido p-toluensulfónico monohidratado y 80 mL de benceno. A continuación, se colocó una trampa Dean Stark y se calentó en baño de aceite a temperatura de reflujo. El calentamiento se mantuvo hasta recolectar 1 mL de agua en el fondo de la trampa, esto duró 3 h. La mezcla se enfrío a temperatura ambiente y se lavó con 2 fracciones de 25 mL de una solución saturada de NaHCO3 y después con 25 mL de salmuera. Se secó con Na2SO4 anhidro y se evaporó el disolvente a presión reducida, obteniéndose 1,617 g (9,5 mmol, 63 %) de un líquido transparente. Sus propiedades espectroscópicas fueron iguales a las encontradas en la literatura24 .
Síntesis de 2,5-dicarbaliloxi-3,4-dihidroxitiofeno 4a
En un baño de hielo se colocó un matraz de fondo redondo de una boca de 25 mL y un agitador magnético. Se agregó 5 mL de alcohol alílico y 143 mg (2 mmol) de etóxido de sodio 95 %. Se agitó fuertemente hasta disolver completamente la sal. Después se agregó 250 µl (0,255 g, 1,5 mmol) de oxalato de dialilo y finalmente se adicionó gota a gota el 0,256 g (1 mmol) del éster 14. La mezcla se agitó por 15 min y después se calentó a reflujo en un baño de aceite durante 4 h, bajo atmósfera de nitrógeno. Se dejó enfriar la mezcla a temperatura ambiente y se filtró el precipitado rojo formado, se lavó con etanol y se secó al vacío durante una noche en un desecador. Los 0,08 g del precipitado que se obtuvieron se disolvieron en 20 mL de agua caliente (60 oC) y se adicionó gota a gota una solución de HCl 2 M hasta pH < 2. El precipitado blanco que se formó se filtró y se secó al vacío durante 24 h. Se obtuvo 0,033 g (0,12 mmol, 12 %) del tiofeno 4a. Polvo blanco (precipitado de agua). p.f. = 108-110 °C. 1H RMN (300 MHz, CDCl3) δ (ppm): 9,28 (s, 2H), 6,04-5,92 (ddd, 2H, J3= 5,6 Hz), 5,44-5,39 (d, 2H, J1= 17 Hz), 5,34-5,31 (d, 2H, J2= 10 Hz), 4,84 -4,81 (d, 4H, J= 5,6 Hz). 13C RMN (CDCl3) δ (ppm): 165,3; 151,9; 131,0; 119,3; 107,1; 66,0. IR ν (cm-1): 3308, 1691, 1669, 1514, 1313, 1214, 1163, 921, 768, 681. AE: encontrado: C=49,93; H=4,07, calculado: C=50,69, H=4,25. EM-IE, [M]= 284; C12H12O6S: 284,04.
Síntesis de 2,5-dicarbaliloxi-3,4-etilendioxitiofeno 2a
En un matraz de fondo redondo de 25 mL de una boca y con agitador magnético se colocó 3 mL de HMPA anhidra; a continuación, se agregaron 0,115 g (0,5 mmol) de ácido 7a (preparado de acuerdo a la referencia 11) y 0,172 g (1,25 mmol) de K2CO3. La mezcla se agitó por 15 min y se adicionó 140 µL (0,252 g, 1,5 mmol) de yoduro de alilo. La reacción se calentó a 80 oC en un baño de aceite durante 2,5 h y bajo atmósfera de nitrógeno. Se dejó enfriar la mezcla a temperatura ambiente y se colocó en un embudo de separación, luego se adicionaron 30 mL de agua y 10 mL de ácido clorhídrico al 10 %. Después se hicieron 2 extracciones de 25 mL con éter. Se juntaron las fracciones etéreas y se lavó con 15 mL de Na2S2O3 al 10 %. La fase orgánica se secó con Na2SO4 anhidro y se evaporó el disolvente a presión reducida en un rotavapor. El residuo se purificó en una columna cromatográfica flash de gel de sílice utilizando como eluyente la mezcla n-hexano:acetato de etilo en orden creciente de polaridad hasta alcanzar la proporción 75:25. Se obtuvieron 0,120 g de un polvo blanco 2a (0,385 mmol, 77 %). p.f.= 133-135 °C. 1H RMN (500 MHz, CDCl3) δ (ppm): 6,03-5,95 (ddd, 2H, J3= 5,5 Hz), 5,44-5,39 (dc, 2H, J1= 17,5, J4= 1,5 Hz), (dc, 2H, J2= 11,5, J4= 1,5 Hz), 4,79 -4,77 (d, 4H, J=5,6 Hz), 4,40 (s, 4H). 13C RMN (125 MHz CDCl3) δ (ppm): 160,4; 145,2; 131,6; 118,7; 111,7; 65,7; 64,7. IR ν (cm-1): 2944, 1694, 1677, 1376, 1304, 1157, 1092, 1021, 765. EM-IE, [M]=310; EM-ES, [M-1]=309,0435, calculado para C14H13O6S: 309,0427; C14H14O6S: 310,05.
Síntesis de 2,6-dimetoxibenzoato de alilo 1
El compuesto 1 se obtuvo por dos métodos diferentes:
Método A: En un matraz de fondo redondo de 25 mL de 1 boca y con agitador magnético se colocó 5 mL de HMPA anhidra a continuación se agregaron 0,560 g (3 mmol) de ácido 9 y 0,620 g (4,5 mmol) de K2CO3. La mezcla se agitó por 15 min y se adicionó 360 µl (0,66 g, 3,9 mmol) de yoduro de alilo. La reacción se calentó a 80 °C en un baño de aceite durante 24 h y bajo atmósfera de nitrógeno. Se dejó enfriar la mezcla a temperatura ambiente y se colocó en un embudo de separación, luego se adicionaron 50 mL de agua y 15 mL de ácido clorhídrico al 10 %. Después se hicieron 2 extracciones de 30 mL con éter. Se juntaron las fracciones etéreas y se enjuagó con 15 mL de Na2S2O3 al 10 %. La fase orgánica se secó con Na2SO4 anhidro y se evaporó el disolvente a presión reducida en un rotavapor. El residuo se purificó en una columna cromatográfica flash de gel de sílice utilizando como eluyente la mezcla n-hexano:acetato de etilo en orden creciente de polaridad hasta alcanzar la proporción 90:10. Se obtuvo 0,398 g (1,8 mmol, 60 %) de un aceite incoloro 1.
Método B: En un matraz de fondo redondo de 1 boca de 25 mL secado a la flama se adicionaron 0,370 g (2 mmol) de 9, 140 µl (0,120 g, 2 mmol) de alcohol alílico y 0,577 g (2 mmol) de trifenilfosfina 97 %. El matraz fue purgado con N2 y fue agregado vía cánula 0,66 mL de tetrahidrofurano (THF). La mezcla (de concentración 3 M en el ácido) fue puesta en un baño sónico de 50-60 Hz y sometida a sonicación durante 10 min, mientras al mismo tiempo se agregaba gota a gota con una jeringa 1,00 mL (0,383 g, 2,2 mmol) de azodicarboxilato de dietilo (DEAD) 40 %. Finalizada la adición, se dejó la mezcla en el baño sónico durante 30 min adicionales. La mezcla resultante fue concentrada a presión reducida en un rotavapor y el residuo fue purificado por columna cromatográfica flash de gel de sílice y utilizando una mezcla de eluyentes de polaridad creciente de n-hexano:acetato de etilo hasta alcanzar la relación 90:10. Se obtuvo 0,266 g (1,2 mmol, 60 %) de un sólido blanco 1. Cristales incoloros (AcOEt) p.f. = 56-57 °C. 1H RMN (300 MHz, CDCl3) δ (ppm): 7,30-7,24 (t, 1H, J= 8,4 Hz), 6,56-6,53 (d, 2H, J= 8,7 Hz) 6,07-5,94 (ddd, 1H, J3= 5,7 Hz), 5,44-5,39 (dc, 1H, J1=17,1 Hz, J4=1,5 Hz), 5,28-5,23 (dc, 1H, J3=10,5 Hz, J4=1,5 Hz), 4,84 -4,81 (dt, 2H, J1=5,7 Hz, J2=1,5 Hz). 13C RMN (75 MHZ, CDCl3) δ (ppm):166,1; 157,3; 132,1; 131,0; 117,8; 113,0; 103,9; 65,7; 55,9. IR ν (cm-1): 2940, 1730, 1594, 1474, 1249, 1105, 1068. AE: encontrado: C=64,61, H=6,07; calculado: C=64,85, H=6,35. EM-IE, [M+H] encontrado 223; C16H20O6S: 222,09.
Resultados y discusión
La reacción de descarboxilación de β-cetoésteres mediada por un catalizador de Pd0 fue probada primero en la molécula 1. Este éster fue sintetizado con un rendimiento de 60 % mediante la reacción del ácido 2,6-dimetoxibenzoico 9 (el cual está disponible comercialmente) con yoduro de alilo y/o alcohol alílico (esquema 2). El ácido 9 ha sido utilizado previamente como molécula modelo en reacciones de descarboxilación mediadas por un catalizador de Pd(II)8 debido a que el carbono ipso se encuentra en posición orto a dos grupos electrodonadores, los mismos que según la evidencia experimental favorecen la eliminación de CO2 cuando se utiliza Pd(CF3COO)2 como catalizador25 .
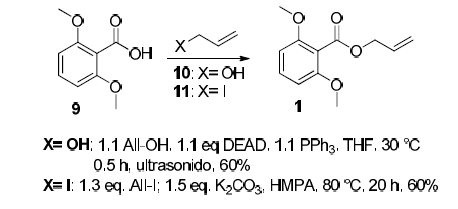
Esquema 2
Síntesis del compuesto 1 por dos métodos
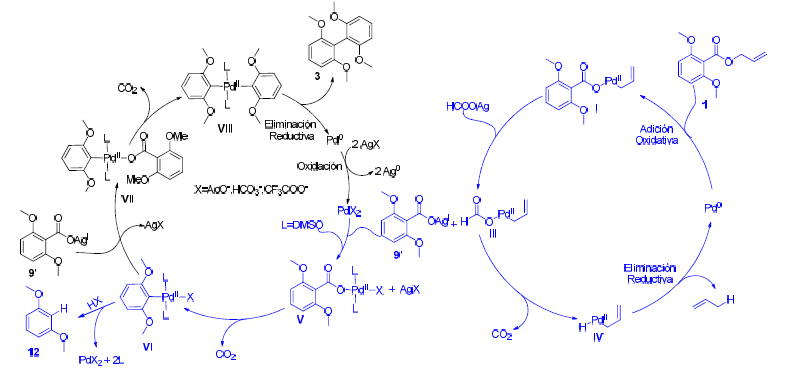
Una vez sintetizado el compuesto 1 se procedió a probar la reacción de descarboxilación. La hipótesis que se manejó fue que el intermediario I (una especie con centro metálico de PdII que resulta por adición oxidativa del catalizador de Pd0 sobre el doble enlace del éster alílico, ver esquema 3) podría sufrir descarboxilación debido al ataque del areno activado al centro metálico formando el intemediario II, en una forma similar a lo que ocurre en las reacciones de descarboxilación en las que se emplea como catalizador Pd(CF3COO)2. En aquel tipo de reacciones, se forman especies de PdII electrofílico, debido a que este metal está coordinado a dos ligantes electroatractores (CF3COO-)25. De esta manera el intermediaro II puede ser protonado por HCOOH.NEt3 generando el producto de descarboxilación 12 y Pd0 una vez que los intermediarios III y IV han sufrido la eliminación de CO2 y pro-peno respectivamente.
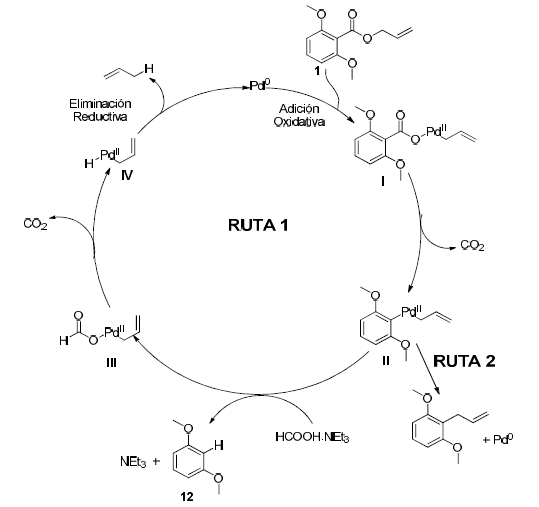
Esquema 3
Hipótesis propuesta para la descarboxilación del éster alílico 1
Los experimentos de descarboxilación del éster 1 se llevaron a cabo a una temperatura de 80 o 110 °C, para favorecer la eliminación de CO2 del éster. Además, en todos los experimentos se utilizó un exceso de HCOOH que actúa como agente reductor, y fuente de protón en la reacción (tabla 1). La reacción se probó con el catalizador Pd[P(Ph)3]4 en una relación del 10 % al sustrato en dos disolventes diferentes tolueno (con NEt3 como base) y DCE (sin ninguna base). El único producto que se obtuvo fue el ácido 9 (tabla 1, ejemplos a-b). A partir del ejemplo c y hasta el ejemplo j se utilizaron catalizadores de PdII para la remoción del grupo alilo. Cabe resaltar que los catalizadores de PdII se reducen a Pd0 en presencia de los ligantes trifenilfosfina y trifenilfosfito. La especie de Pd0 formada a partir de esta reacción es la que se incorpora al ciclo catalítico. En primer lugar, se evaluó el catalizador Pd(OAc)2 con ligantes fosfina y fosfito, en DMF como disolvente (tabla 1, ejemplos c-e). Tampoco se consiguió el compuesto 12 con este catalizador. En los experimentos de a-e, se utilizó 10% mol de catalizador en relación al sustrato de acuerdo al procedimiento informado por Tunge20 para la descarboxilación-alilación de ésteres alílicos de ácidos acetilénicos. En esta serie de experimentos se evidenció la desprotección del grupo carboxilato.
En reacciones de acoplamiento descarboxilativo mediadas por sales de PdII, se utiliza con frecuencia la base Ag2CO3 . En ellas, el correspondiente carboxilato de plata (I) (de areno o heteroareno) reacciona con la sal de PdII por intercambio de anión, formando una especie de PdII con dos diferentes aniones carboxilato. La especie en cuestión sufre eliminación de CO2, resultando en un intermediario de areno-PdII, el mismo que está coordinado a DMSO que actúa como ligante y que se adiciona cuando empieza la reacción. Por lo tanto, en los demás ejemplos, el Ag2CO3 fue utilizado en vez de NEt3 y DMSO (al 5 % en volumen) como segundo ligante. Estas reacciones fueron monitoreadas por 20 h y los resultados de esta estrategia se muestran en la tabla 1 (ejemplos f-j). En los experimentos f-h se utilizó un exceso de sal de PdII con respecto al ligante trifenilfosfina, de manera que el exceso de sal de PdII sirva como segundo catalizador una vez formado el carboxilato de plata. Además, se adicionó un exceso de Ag2CO3 para asegurar la formación in situ de formiato de plata (I). Tanto la sal de Pd como la de Ag fueron adicionadas al inicio de la reacción. En estos casos el ácido 2,6dimetoxibenzoico se obtuvo como producto principal.
Además, se observó que el rendimiento de reacción es sensible a la cantidad de base utilizada (ejemplo g, tabla 1). La reacción de reducción que sufre el catalizador de PdII (a pesar del exceso) por el ligante trifenilfosfina parece ser una reacción competitiva, la cual afecta directamente la formación de especies activas de descarboxliación y por lo tanto, impide que se efectúe la reacción deseada. Tomando en cuenta lo anterior, en los ejemplos i y j se intentó la reacción, adicionando un exceso de Pd(CF3CO2)2 una vez que se ha llevado a cabo la formación del 2,6-dimetoxibenzoato de plata (I) 9’, que se estimó en 2 h de acuerdo al ejemplo a. Además, para que la trifenilfosfina no reduzca el nuevo catalizador adicionado se agregaron 2 h después de comenzada la reacción dos aditivos, DIAD en el experimento i y CBr4/alcohol alílico, en el experimento j. En ambos casos se agregaron antes que la cantidad extra del catalizador de PdII. De esta forma al usar DIAD como aditivo se obtuvieron los compuestos 12 y 3 en 5 % y 47 % de rendimiento, esto significa un rendimiento ligeramente superior a 50 % de productos de descarboxilación. El compuesto de homoacoplamiento 3 ha sido obtenido previamente por medio de una metodología tipo Ullman por el grupo de Koning durante la síntesis de quinonacardinalina 3 un compuesto biológicamente activo29 .
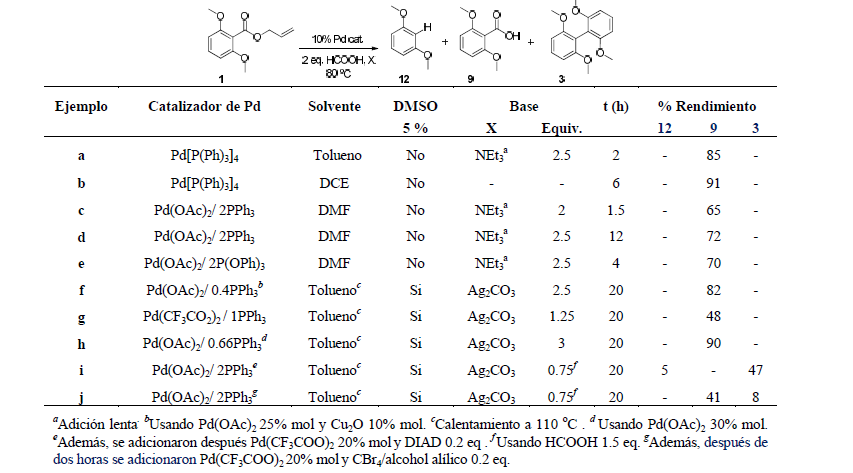
a Adición lenta. b Usando Pd(OAc)2 25% mol y Cu2O 10% mol. c Calentamiento a 110 °C . d Usando Pd(OAc)2 30% mol. e Además, se adicionaron después Pd(CF3COO)2 20% mol y DIAD 0.2 eq . f Usando HCOOH 1.5 eq. g Además, después de dos horas se adicionaron Pd(CF3COO)220% moly CBr4/alcohol alílico 0.2 eq.
Además, La molécula biarílica 3 no se aisló durante el desa-biarílico como producto de descarboxilación en bajo rendirrollo de la metodología de homo y heteroacoplamiento des-miento, y con el ácido 9 como producto mayoritario. Finalcarboxilativo mediado por sales de PdII informado por el gru-mente, para demostrar que el intermediario de PdIIIIno es lo po de Deng27. Esta es la primera vez que se informa una reacción suficientemente electrofílico para sufrir una reacción de deshomoaclopamiento aril-aril a partir de un éster alílico carboxilación, se efectuó un experimento en ausencia de de ácido benzoico. Con el otro aditivo CBr4/alcohol alílico se fuente de protón, en el que 2 h después de empezada la reacción obtuvo el sistema se observó la precipitación de un polvo negro que se atribuye a Pd elemental. La formación de los productos 3 y 12 se racionalizó en el esquema 4, inspirados en los trabajos de Tsuji19 y Larrosa26 .
Esquema 4
Mecanismo propuesto para la descarboxilación del éster alílico 1
El éster alílico 1 en presencia de un catalizador de Pd0 sufre adición oxidativa para formar el intermediario I, el cual produce benzoato de plata (I) 9’ y el intermediario III (el mismo que después de eliminar CO2 y propeno, genera el catalizador de Pd0) en la presencia de formiato de plata. La especie 9’ puede ser protonada y generar el ácido 2,6-dimetoxibenzoico 9 (ejemplo j, tabla 1). La especie 9’ sufre una reacción de transmetalación con la sal de PdII para producir el intermediario V. El cual experimenta eliminación de CO2 para originar la especie aril-Pd VI, la cual puede ser protonada por especies ácidas y producir el compuesto 12. Además, el intermediario VI puede sufrir intercambio de anión con el carboxilato de plata (I) 9’ dando lugar al intermediario VII, el cual sufre eliminación de CO2 dando origen a la especie VIII. Este intermediario experimenta eliminación reductiva para generar el compuesto 3 y una especie de Pd0, el mismo que en presencia de especies de plata (I) puede ser oxidada a sales de Pd(II) que sirven como catalizador en la reacción de descarboxilación.
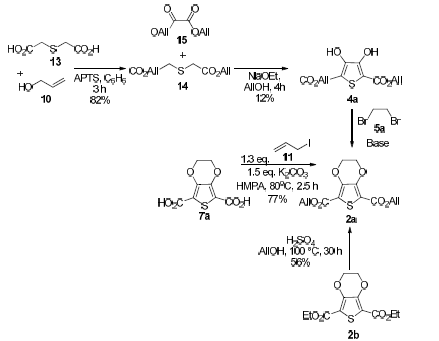
Los resultados alcanzados en la producción del compuesto de descarboxilación 12 a partir del éster alílico 1, demostraron que la descarboxilación del heterociclo 2a podía ser factible, en condiciones relativamente más suaves si comparamos con las metodologías basadas catalizadores de Cu2-5 y/o Ag6-7 . Como se mencionó anteriormente la eliminación de CO2 mediada por catalizadores de PdII es favorecida cuando el compuesto areno está sustituido por grupos electrodonadores. De acuerdo al mecanismo propuesto para la descarboxilación de ácidos aromáticos mediada por sales de PdII8, en el benzoato coordinado al centro metálico, el fragmento de areno activado ataca al metal PdII electrofílico en forma similar a lo que ocurre en una sustitución electrofílica aromática. Además, el PdII está coordinado a ligantes DMSO. Trasladando esto para el caso del anillo de tiofeno, la posición α sufre reacciones de sustitución electrofílica con mayor facilidad que los sistemas areno30. Además, en el caso de los 3,4dialcoxitiofenos esta posición está muy activada por el efecto electrónico del sustituyente alcóxido como ha sido demostrado en la electropolimerización del etilendioxitiofeno31 . Se pensó que estos dos factores podrían favorecer la descarboxilación en las moléculas tipo 2. Para probar esta idea, como primer paso era necesario obtener eficientemente el éster 2a. La ruta sintética hacia este compuesto requiere la preparación del compuesto 2,5-dicarbaliloxi-3,4-dihidroxitiofeno 4a, el cual después de la reacción de O-alquilación con 1,2dibromoetano proporcionaría el compuesto deseado 2a. El primer intento para preparar eficientemente el compuesto 4a fue por medio de la reacción de Hinsberg (esquema 5), que es la metodología preferida para obtener ésteres del ácido 2,5dicarboxílco-3,4-dihidroxitiofeno32. La primera reacción entre el ácido tiodiglicólico 13 y alcohol alílico 10 tuvo un rendimiento del 82%, cuando se empleó una trampa de Dean-Stark. Sin embargo, en el paso de condensación con el oxalato esta metodología falla. Cuando la reacción se efectúa con oxalato de etilo se obtiene como subproducto de reacción el 2,5-dicarbetoxi-3,4-dihidroxitiofeno. El uso de sodio metálico como base, da lugar al mismo subproducto. El tiofeno 4a fue obtenido como un solo producto con un rendimiento del 12 % cuando se utilizó el oxalato de dialilo 13. Dos métodos alternativos para preparar la molécula 2a fueron ensayados (esquema 5). El primero consiste en la alquilación del ácido 2,5dicarboxílico-3,4-etilendioxitiofeno 7a con yoduro de alilo. El compuesto 7a puede ser sintetizado con buen rendimiento de acuerdo a lo informado en la literatura11. La reacción de Oalquilación de 7a tuvo un rendimiento del 77 % cuando se llevó a cabo con el disolvente HMPA. El segundo ensayo consistió en una reacción de transesterificación del compuesto 2b en alcohol alílico utilizando H2SO4 como catalizador, esto permitió la conversión de 2b en 2a en 56 % luego de 30 h de reflujo.
Esquema 5
Rutas de síntesis del ester 2a
La reacción de descarboxilación del compuesto 2a se llevó a cabo en forma similar a lo ocurrido con el éster 1, pero no se logró aislar 3,4-etilendioxitiofeno 8a u otro producto de descarboxilación. La falta de reactividad en el sistema de tiofeno puede ser atribuida a la baja solubilidad de las especies carboxilato en tolueno y/o al bajo carácter electrofílico del catalizador de paladio.
La difenil-(2,3,4,5-tetrafluorofenil)fosfina es un ligante que ha sido probado en la reacción de acoplamiento aril-alquil de Negishi con catalizador de paladio, y no se obtuvo el correspondiente producto de acoplamiento. Al contrario, se obtuvo eficientemente un producto de deshalogenación del halogenuro de arilo. En otras palabras, este ligante evitó la eliminación reductiva en especies tipo II (ruta 2, esquema 4)33. Por lo tanto, parece plausible utilizar esta fosfina electroatractora confiera el carácter electrofílico al centro metálico en una hipotética especie de paladio tipo I (esquema 3), el mismo que podría ser atacado por un fragmento areno o heteroareno activado, produciendo un intermediario tipo II. Esta especie de PdII podría ser protonada por ácido fórmico como fue propuesto para el intermediario II (esquema 3) y, de esta manera se podría evitar el uso de Ag2CO3 como base y aditivo.
Conclusiones
Se demostró que la descarboxilación directa del éster alílico del ácido 2,6-dimetoxibenzoico es posible cuando la reacción se realiza con combinación de catalizadores Pd(OAc)2/PPh3 10 % mol y Pd(CF3COO)2 20 % en tolueno con DMSO al 5 %, uitlizando 1,5 eq HCOOAg y 0,2 eq de DIAD como aditivos. En estas condiciones de reacción fue posible tener dos productos de descarboxilación 12 y 3 en un rendimiento global de 52 %, en un proceso que implica al menos dos reacciones consecutivas en el mismo matraz. El procedimiento podría ser mejorado y probado en la síntesis de sistemas biarílicos con impedimento estérico. Un catalizador diseñado para los requerimientos electrónicos descritos podría mejorar el desempeño de la reacción. La aplicación de este proceso como tal en la síntesis de 3,4-dialcoxitiofenos requiere un estudio más profundo, pero los resultados mostrados abren la posibilidad de desarrollar una ruta sintética con otro catalizador o bajo condiciones de reacción diferentes.
Descarboxilación de dos ésteres alílicos de ácidos areno carboxílicos mediada por catalizadores de paladio, hacia la síntesis de 3,4-dialcoxitiofenos
1. LJ Goossen, G Deng, G.; LM Levy. Synthesis of biaryls via catalytic decarboxylative coupling. Science, 313, 662-664 (2006).
2. A Cairncross, JR Roland, RM Henderson, WA Sheppard. Organocopper intermediates via decarboxylation of cuprous carboxylates. J. Am. Chem. Soc., 92, 3187-3189, (1970).
3. T Cohen, RA Schambach. Copper-quinoline decarboxylation. J. Am. Chem. Soc., 92, 3189-3190 (1970).
4. T Cohen, RW Berninger, JT Wood. Products and kinetics of decarboxylation of activated and unactivated aromatic cuprous carboxylates in pyridine and in quinoline. J. Org. Chem., 43, 837-848 (1978).
5. LJ Goossen, WR Thiel, N Rodríguez, C Linder, B Melzer. Copper-catalyzed protodecarboxylation of aromatic carboxylic acids. Adv. Synth. Catal., 349, 2241-2246 (2007).
6. LJ Goossen, C Linder, N Rodriguez, PP Lange, A Fromm. Sil-ver-catalysed protodecarboxylation of carboxylic acids. Chem. Commun., 46, 7173-7175 (2009).
7. P Lu, C Sanchez, J Cornella, I Larrosa. Silver-catalyzed protodecarboxylation of heteroaromatic carboxylic acids. Org. Lett., 11, 5710-5713 (2009).
8. JS Dickstein, CA Mulrooney, EM O'Brien, BJ Morgan, MC Kozlowski. Development of a catalytic aromatic decarboxylation reaction. Org. Lett., 9, 2441-2444 (2007).
9. Z Shen, Z Ni, S Mo, J Wang, Y Zhu. Palladium-catalyzed intramolecular decarboxylative coupling of arene carboxylic acids/esters with aryl bromides. Chem. Eur., 18, 4859-4865 (2012).
10. JJ Dai, JH Liu, DF Luo, L Liu. Pd-catalysed decarboxylative Suzuki reactions and orthogonal cu-based O-arylation of aromatic carboxylic acids. Chem. Commun., 47, 677-679 (2011).
11. PA Cisneros-Pérez, D Martínez-Otero, E Cuevas-Yáñez, BA Uribe-Frontana. Diprotodecarboxylation Reactions of 3,4-Dialkoxythiophene-2,5-dicarboxylic Acids Mediated by Ag2CO3 and Microwaves. Synth. Commun., 44, 222-230 (2014).
12. SA Mauger, AJ Moulé. Characterization of new transparent organic electrode materials. Org. Electron., 12, 1948-1956 (2011).
13. Q Yan, Y Zhou, BB Ni, Y Ma, J Wang, J Pei, Y Cao. Organic semiconducting materials from sulfur-hetero benzo[k]fluoranthene derivatives: Synthesis, photophysical properties, and thin film transistor fabrication. J. Org. Chem., 73, 5328-5339 (2008).
14. AL Dyer, EJ Thompson, JR Reynolds. Completing the color palette with spray-processable polymer electrochromics. ACS Appl. Mater. Interfaces, 3, 1787-1795 (2011).
15. N Rozlosnik. New directions in medical biosensors employing poly(3,4-ethylenedioxy thiophene) derivative-based electrodes. Anal. Bioanal. Chem., 395, 637-645, (2009).
16. RMF Batista, E Oliveira, C Nuñez, SPG Costa, C Lodeiro, MMM Raposo. Synthesis and evaluation of new thienyl and bithienyl-bis-indolylmethanes as colorimetric sensors for anions. J. Phys. Org. Chem., 22, 362-366 (2009).
17. PM Beaujuge, J Subbiah, KR Choudhury, S Ellinger, TD McCarley, F So, JR Reynolds. Green dioxythiophenebenzothiadiazole donor−acceptor copolymers for photovoltaic device applications. Chem. Mater., 22, 2093-2106 (2010).
18. PA Cisneros-Pérez. Tesis Doctoral, Universidad Nacional Autónoma de México, 2014.
19. J Tsuji, M Nisar, I Shimizu. Facile palladium-catalyzed decarboxylation reaction of allylic beta-keto esters. J. Org. Chem., 50, 3416-3417 (1985).
20. DK Rayabarapu, JA Tunge. Catalytic decarboxylative sp−sp3 coupling. J. Am. Chem. Soc., 127, 13510-13511 (2005).
21. R Jana, R Trivedi, JA Tunge. Mild decarboxylative allylation of coumarins. Org. Lett., 11, 3434-3436 (2009).
22. A Kumar, DM Welsh, MC Morvant, F Piroux, KA Abboud, JR Reynolds. Conducting poly(3,4-alkylenedioxythiophene) derivatives as fast electrochromics with high-contrast ratios. Chem. Mater., 10, 896-902 (1998).
23. DM Welsh, A Kumar, EW Meijer, JR Reynolds. Enhanced contrast ratios and rapid switching in electrochromics based on poly(3,4-propylenedioxythiophene) derivatives. Adv. Mater., 11, 1379-1382 (1999).
24. D Imao, A Itoi, A Yamazaki, M Shirakura, R Ohtoshi, K Ogata, Y Ohmori, T Ohta, Y Ito. Easy access to esters with a benzylic quaternary carbon center from diallyl malonates by palladiumcatalyzed decarboxylative allylation. J. Org. Chem., 72, 1652-1658 (2007).
25. D Tanaka, SP Romeril, AG Myers. On the mechanism of the palladium(II)-catalyzed decarboxylative olefination of arene carboxylic acids. Crystallographic characterization of nonphosphine palladium(II) intermediates and observation of their stepwise transformation in Heck-like processes. J. Am. Chem. Soc., 127, 10323-10333 (2005).
26. J Cornella, H Lahlali, I Larrosa. Decarboxylative homocoupling of (hetero)aromatic carboxylic acids. Chem. Commun., 46, 8276-8278 (2010).
27. K Xie, S Wang, Z Yang, J Liu, AWang, X Li, Z Tan, CC Guo, W Deng. Synthesis of Biaryls by Pd-Catalyzed Decarboxylative Homo-and Heterocoupling of Substituted Benzoic Acids. Eur. J. Org. Chem., 49, 5787-5790 (2011).
28. K Xie, Z Yang, X Zhou, X Li, S Wang, Z Tan. X An, CC Guo. Pd-catalyzed decarboxylative arylation of thiazole, benzoxazole, and polyfluorobenzene with substituted benzoic acids. Org. Lett., 12, 1564-1567 (2010).
29. S Govender, EM Mmutlane, WAL van Otterlo, CB de Koning. Bidirectional racemic synthesis of the biologically active quinonecardinalin 3. Org. Biomol. Chem., 5, 2433-2440 (2007).
30. T Eicher, S Hauptmann. The Chemistry of Heterocycles. Wiley-VCH, Weinheim, 2003.
31. M Dietrich, J Heinze, G Heywang, F Jonas. Electrochemical and spectroscopic characterization of polyalkylenedioxythiophenes. J. Electroanal. Chem., 369, 87-92 (1994).
32. A Kumar, BD Tilak. Ester of thiodiglycollic & thiopropionic acids & 2,5-dicarboxy-3,4-dihydroxythiophene as potential slow activity anticancer agents. Indian J. Chem., 25B, 880-882 (1988).
33. E Gioria, JM Martínez-Ilarduya, D García-Cuadrado, JA Miguel, M Genov, P Espinet. Phosphines with Tethered Electron-Withdrawing Olefins as Ligands for Efficient Pd-Catalyzed Aryl-Alkyl Coupling. Organometallics, 32, 4255-4261 (2013).