Variabilidad Genética de Líneas Endogámicas de Maíz Comparadas con Progenitores Criollos mediante Microsatélites
Genetic Variability of Maize Inbred Lines Compared with Parental Landraces by Means of Microsatellite Markers
Variabilidad Genética de Líneas Endogámicas de Maíz Comparadas con Progenitores Criollos mediante Microsatélites
Conciencia Tecnológica, núm. 51, pp. 47-52, 2016
Instituto Tecnológico de Aguascalientes
Recepción: 29 Agosto 2013
Aprobación: 07 Diciembre 2015
Financiamiento
Fuente: PRODEP
Nº de contrato: IDCA: 9869. CLAVE: ITROQ-CA-2
Beneficiario: Instituto Tecnológico de Roque.
Resumen: El conocimiento de la diversidad genética en líneas puras de maíz (Zea mays L.), un cereal básico y el más importante en la dieta de los mexicanos es indispensable en un programa de mejoramiento genético. En la actualidad existe un déficit productivo el cual puede ser reducido con la obtención de mejores genotipos de maíz. Por lo tanto, el objetivo de esta investigación fue seleccionar una colección de marcadores moleculares tipo microsatélites repetidos de secuencia corta (SSRs) relacionados con características deseables de líneas endogámicas de maíz generadas a partir de una colección inicial de maíces criollos, y analizar la diversidad genética de la población original y de las líneas endogámicas. De una población inicial de 600 genotipos de maíz derivados en el Instituto Tecnológico de Roque con buenas características agronómicas, se eligieron 80 líneas endogámicas S1, las que se evaluaron durante el ciclo agrícola 2010 para seleccionar genotipos que superaran en rendimiento a los progenitores. Solo siete líneas endogámicas S2 derivadas de este ensayo fueron evaluados en esta investigación, para continuar con el proceso de endogamia. La variabilidad genética detectada muestra la presencia de polimorfismos y una amplia distribución de genotipos en la población, lo que indica la posibilidad de continuar con el proceso de identificación de líneas superiores mediante selección para la explotación de heterosis.
Palabras clave: maíces criollos, líneas endogámicas, diversidad genética, SSR.
Abstract: Knowledge of genetic diversity in maize (Zea mays L.) inbred lines, a basic cereal and the most important in Mexican population diet, is indispensable in a breeding program. Nowadays there is a production deficit which can be reduced by achieving better genotypes of maize. Therefore, the objective of this research was to select a collection of microsatellite molecular markers type Simple Sequence Repeats (SSRs) associated with desirable traits of maize inbred lines generated from a collection of native maize, and analyze the genetic diversity of the original population and the derived inbred lines. 80 inbred lines S1 were evaluated during the 2010 agricultural cycle to select genotypes with higher yielding ability than parental lines from an initial population of 600 genotypes of maize derived at Instituto Tecnológico de Roque with good agronomic traits. From this trail, only seven S2 inbred lines were evaluated in this research to continue the inbreeding process. The detected genetic variability showed the presence of polymorphisms and a wide distribution of genotypes in the population, suggesting the possibility of selecting top lines for heterosis exploitation.
Keywords: maize landraces, inbred lines, genetic diversity, SSR.
Introducción
La generalización de las técnicas de Biología Molecular ha tenido, entre otras, dos consecuencias trascendentales [1]: Por una parte el desarrollo de una poderosa herramienta de uso general para el análisis genético tanto en estudios básicos como aplicados, los marcadores moleculares. Por otra una visión mucho más completa de la variabilidad genética y su distribución a distintos niveles: individual, población y especie. El uso de tales herramientas y los nuevos datos adquiridos con ellas están teniendo profundas repercusiones teóricas y prácticas.
El mejoramiento de poblaciones de maíz por métodos convencionales se caracteriza por ser un proceso lento y sin información detallada acerca de la evolución de los caracteres seleccionados; por ello, los genotipos liberados en ocasiones quedan desfasados en relación a las condiciones del campo; además, existen dificultades en la caracterización y protección intelectual de las variedades generadas [2]. Se genera un desconocimiento de los caracteres secundarios que cosegregan en las selecciones y los programas nacionales resultan con baja competitividad frente a las empresas trasnacionales de producción de semilla.
Cada vez es más apremiante la necesidad de obtener variedades a corto plazo, más adaptadas a las condiciones ambientales del sistema de temporal regular a bueno con una entrada mínima de insumos e incrementar el potencial productivo de los agro-ecosistemas. Actualmente el proceso de liberación de variedades mejoradas para condiciones de temporal es muy lento debido a la poca eficiencia de la selección por efecto del medio ambiente. La aplicación de las tecnologías derivadas de la biología molecular permitirá acelerar el proceso de liberación de genotipos. Cabe señalar que a la fecha es poca la interacción que existe entre los fitomejoradores tradicionales y biotecnólogos. Esta interacción es necesaria para alimentar a la población en constante crecimiento y adaptar las prácticas agrícolas de cara al cambio climático. Al acortar los plazos de selección y liberación de variedades se logrará un ahorro sustancial de recursos económicos, humanos y ambientales.
Las empresas trasnacionales no se ocupan de la mejora de plantas para la agricultura de temporal y las instituciones nacionales que lo hacen están limitadas por la disponibilidad de recursos y de personal. Como se ha señalado parece haber falta de interacción entre los laboratorios que hacen biología molecular, proteómica, metabolómica y secuenciación de genomas y los fitomejoradores tradicionales. Es necesario pues establecer el nexo y la colaboración para que los resultados obtenidos en los laboratorios que usan tecnologías de punta puedan ser aprovechados en desarrollo efectivo del campo mexicano. Con este proyecto se pretende avanzar conjuntamente en la selección de líneas mediante criterios fenotípicos, pero también incluyendo una caracterización a nivel molecular y bioquímico.
Entre los cereales más importantes, se encuentra el maíz, que es el cultivo más importante en México, desde el punto de vista alimentario, industrial, político y social; su importancia radica en la amplia utilización para la alimentación humana y animal. Este cultivo se utiliza como grano en la alimentación humana, como materia prima en la industria y la planta puede emplearse como forraje verde, ensilado, rastrojo, forraje fresco y materia orgánica al suelo; finalmente como mazorca tierna (elote) se consume como alimento fresco. Actualmente, uno de los usos más sobresalientes es que el grano se está empleando para obtener biocombustibles [3], práctica la cual ha sido prohibida en México por representar una amenaza a la soberanía alimentaria del país. En varios países se sigue usando maíz como materia prima para producir etanol, lo que supone algunos inconvenientes. El principal, el más criticable desde el punto de vista moral, es que produce un aumento en el precio de los alimentos, ya que hay una menor disponibilidad de una materia esencial, como es el maíz.
El maíz mexicano padece las consecuencias de una baja productividad, escenario que ha permitido un aumento sostenido en las importaciones del grano, que alcanzaron en el 2011 un monto superior a 10 millones de toneladas. El valor de las importaciones del producto alimenticio más importante para el campo nacional, atendiendo a su valor histórico y comercial, superó los 2,769 millones de dólares el año pasado, siendo Estados Unidos el principal proveedor del país y líder global en la siembra del cultivo [4].
Actualmente se han liberado muy pocas variedades mejoradas para condiciones de temporal regular a bueno que permitan aumentar significativamente el rendimiento unitario. La producción de este cultivo depende muchas veces de genotipos criollos con bajo potencial de rendimiento y algunas características agronómicas no favorables para cubrir los requerimientos de la agricultura moderna, o bien, depende de semillas importadas no adaptadas a las condiciones ambientales regionales, aunado a los precios altos que limitan la accesibilidad por parte del agricultor de escasos recursos económicos [5]. Por esta razón el objetivo de esta investigación fue seleccionar una colección de marcadores moleculares tipo microsatélites (SSR) relacionados con características deseables de líneas endogámicas de maíz generadas a partir de una colección inicial de maíces criollos.
Fundamentos teóricos
Existen varias definiciones de marcador genético. Gale [6] lo define como cualquier medio para identificar cualquier locus específico en un cromosoma. En definitiva podemos usar como marcador el efecto de un gen fácilmente observable en los individuos (genéricamente conocidos como caracteres morfológicos), metabolitos característicos de bajo peso molecular, proteínas que puedan extraerse y observarse con facilidad (isoenzimas, proteínas de reserva y proteínas del suero) generalmente tras un fraccionamiento mediante electroforesis o segmentos de DNA que pueden obtenerse e identificarse por toda una serie de técnicas moleculares [7].
Los marcadores bioquímicos y moleculares se han usado extensivamente en el estudio de la estructura genética de poblaciones y de la evolución [7] y se han usado como herramientas en fitomejoramiento [1, 8]. El uso de isoenzimas permitió que por primera vez se pudiese elegir un grupo de genes sin saber previamente si eran variables o no, o su grado de variabilidad, en el nivel taxonómico a estudiar. Los genes de isoenzimas representaban por tanto un grupo de genes que podía considerarse representativo del acervo genético de una población o especie, es decir la información genética total codificada en la suma total de sus genes en un momento dado, que permitía una estima correcta de la variabilidad total, o lo más próximo a ello [9]. Los marcadores moleculares tienen la ventaja de ser fenotípicamente neutros, presentan codominancia y muchos polimorfismos, pueden ser evaluados en cualquier estado de desarrollo de la planta y en cualquier época del año sin que haya interferencia de efectos epistáticos o epigenéticos. Dentro de estos, los SSR son regiones de secuencias pequeñas (2-10 bases) repetidas arregladas en serie, que se asume están distribuidas por todo el ADN. Son altamente variables y mutables, frecuentemente presentan gran variación y esto permite usarlas para medir el polimorfismo entre especies y variedades relacionadas [10, 2]. Los SSR pueden permitir medir la diversidad y asignar grupos heteróticos o usarse para obtener huellas genéticas con igual o mayor eficiencia y menores costos que, por ejemplo, los marcadores genéticos tipo RFLPs [11].
Gaut y colaboradores y Tenaillon y colaboradores [13, 14] realizaron experimentos para explorar a fondo la variación genética a nivel de secuencias en el maíz. Como ya se mencionó, los niveles de variación genética son muy altos en esta especie. El tamaño del genoma del maíz se ha expandido a 2.3 gigabases, aproximadamente, durante los últimos 3 millones de años mediante la proliferación de retrotransposones con repeticiones de terminales largas (retrotransposones LTR). En este apartado destaca el hecho de que casi el 85% del genoma del maíz está compuesto de cientos de familias de elementos de transposición, dispersos de forma no uniforme a través de todo el genoma [12].
El maíz en México fue domesticado a partir del teosintle, un pasto silvestre, hace aproximadamente 6,300 años. Después de la domesticación inicial, los primeros agricultores continuaron con la selección sobre características morfológicas y bioquímicas benéficas en este importante cultivo. Se han analizado tres genes: uno en el control de la arquitectura de la planta, otro en la síntesis de proteínas de reserva y uno más en la síntesis de almidón, empleando muestras arqueológicas de maíz de México y el suroeste de Estados Unidos. Los resultados revelan que los alelos típicos de maíz contemporáneo estuvieron presentes en el maíz mexicano desde hace 4,000 años [15]. Los datos recientes indican que el maíz fue domesticado entre los estados de Guerrero y Michoacán, en la cuenca del Río Balsas [16].
Materiales y métodos
El trabajo se llevó a cabo en el laboratorio de Sanidad de Semillas y Biología Molecular, de la División de Estudios de Posgrado e Investigación del Instituto Tecnológico de Roque, Celaya, Gto.
De 600 genotipos de maíz, provenientes de los estados de Morelos, Michoacán y Puebla, seleccionados por sus características agronómicas sobresalientes, se derivaron familias de medios hermanos. Se determinó que 169 familias fueron las más sobresalientes y se derivaron líneas endogámicas de primera generación S1 por autofecundación durante el ciclo 2009 y de éstas se seleccionaron 80 en el ciclo 2010 que se evaluaron para seleccionar genotipos que superaran en rendimiento a los maíces criollos [17]. A partir de esta evaluación, se derivaron siete líneas endogámicas de segunda generación S2 por autofecundación, las que se evaluaron para continuar con el proceso de formación de líneas endogámicas y comenzar a formar híbridos. Una vez obtenido el material biológico, se procedió a realizar la extracción de ADN del mismo.
Las líneas estudiadas fueron: Original, Línea Roque Rosas 1, Línea Roque Rosas 2, Línea 8, Línea 17, Línea 65, Línea 73 y Línea 81.
La extracción de ADN se realizó conforme a Liu [18]. Se pusieron a germinar semillas de las líneas S2 seleccionadas en cajas de Petri para la producción de plántulas y obtener tejido vegetal. El tejido se congeló con nitrógeno líquido y se molió en un mortero. A la muestra obtenida se le agregaron 400 L de buffer de extracción de ADN de tejido vegetal (Tris 50 mM, NaCl 300 mM, EDTA 20 mM, SDS 0.5%, Sarkosyl 2%, urea 5M) [18]. Se mezcló utilizando agitador vortex (Genie MR ) y al finalizar, se realizó limpieza con fenol-cloroformo. La fase acuosa se transfirió a un tubo nuevo y se adicionaron dos volúmenes de etanol absoluto pre-enfriado a -20 ˚C. Se mezcló por inversión y se incubó 30 min a -20 ˚C. Se centrifugó por 5 min a 13200 rpm en microcentrífuga (Eppendorf MR 5415D). Se descartó el sobrenadante y se lavó la pastilla con etanol al 70%. Se eliminó el sobrenadante y se secó la pastilla y se suspendió la pastilla en agua desionizada.
El ADN extraído de las plántulas fue tratado con RNasa A (12 L de una solución de 10 g/ L, por 2h a 37 ºC); este ADN así como los productos obtenidos de todas las reacciones llevadas a cabo fueron confirmados mediante electroforesis en gel de agarosa al 2%. El marcador de peso molecular empleado fue Generuler 100 (bp) DNA Ladder de Fermentas LifeSciences MR Los geles se sometieron a electroforesis a 85 Volts. Para la visualización de las bandas, los geles fueron teñidos con bromuro de etidio y fueron observados en transiluminador ultravioleta.
Amplificación de SSR . La matriz base para la generación del dendrograma se construyó con el resultado de las reacciones de PCR empleando un conjunto de oligonucleótidos SSR de maíz (SIGMAALDRICH MR, No. Cat. M 4193); se seleccionó un conjunto de 34 pares de oligonucleótidos dispersos en el genoma de maíz. Los pares de iniciadores fueron seleccionados para cubrir el genoma completo del maíz con un promedio de 20 cM, unidades de distancia en el mapa entre dos marcadores SSR.
Una vez reconstituidos conforme al instructivo del proveedor, se prepararon soluciones de trabajo a una concentración de 2.5 M.
Se realizaron amplificaciones en tubos de microcentrífuga de 0.5 ml, se mezclaron a una concentración final de los componentes mostrados en el Cuadro 1.
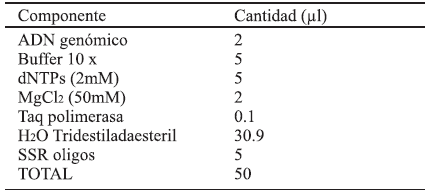
El pH del buffer de reacción fue de 8.3 medido a 25 ˚C. La amplificación de los ácidos nucleicos se llevó a cabo en termociclador Techne MR utilizando el programa de temperaturas que se enlista en el Cuadro 2.
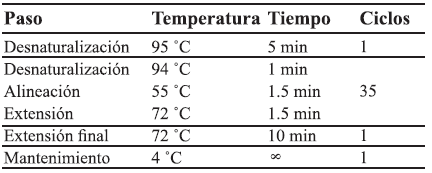
Se tomó una muestra de 13 l para analizar el producto de PCR por electroforesis en gel de agarosa al 2%, tiñendo el gel con bromuro de etidio.
Para cada reacción de amplificación se registró la presencia o ausencia de bandas amplificadas en una matriz binaria codificada en cero (ausencia) y uno (presencia), para detectar los marcadores polimórficos y realizar el análisis.
El análisis de conglomerados es una técnica multivariada que busca agrupar elementos (o variables) tratando de lograr la máxima homogeneidad en cada grupo y la mayor diferencia entre los grupos. A partir de la matriz binaria, se generó un dendrograma mediante el programa NTSYS ver. 2 alimentando los datos dentro del mismo.
Con el empleo del algoritmo UPGMA (Unweighted Pair Group Method Average) y el coeficiente de Jaccard, dentro del programa, se generó un dendrograma de la forma en que se agrupan las líneas estudiadas en base a los SSR [19].
Resultados
El polimorfismo se encuentra presente en la gran mayoría de los oligos en cada una de las muestras, lo que indica que existe una gran variabilidad entre los mismos. Esto es un resultado esperado, al tener un grado de homocigosis bajo al ser líneas S2.
Como se observa en el dendrograma de la figura 1, las líneas con mayor grado de similitud son la L8 con la L81 y RR1 con RR2, con coeficientes de 0.768 y 0.765, respectivamente, mientras que, por lo contrario, entre la L8 y L65 el coeficiente es de tan solo 0.471, lo que representa el valor de similitud más bajo.
En la Figura 1 se puede observar el fenograma obtenido utilizando el método de UPGMA. La agrupación de los materiales muestra la forma en la que se encuentra la distribución entre las líneas y la población original. De esto se puede inferir una relación entre las mismas.
La variabilidad fenotípica es uno de los obstáculos más grandes para el mejoramiento genético convencional. En los ensayos de campo siempre hay mucha variabilidad. La influencia predominante del ambiente representa un problema, ya que genotipos iguales en ambientes diferentes pueden tener fenotipos muy diferentes, mientras que genotipos diferentes en un mismo ambiente pueden tener fenotipos muy parecidos. Es decir, el factor del ambiente puede enmascarar a los factores genéticos. Las Líneas 8 y 81 tienen un grado de similitud alto, lo que llevaría a pensar que difieren en pocos marcadores.
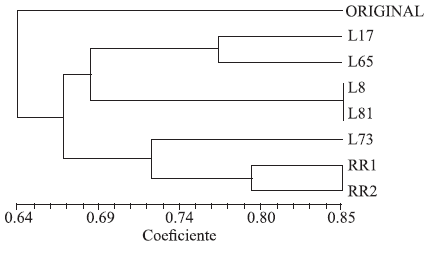
Figura 1.
Dendrograma de similitud de las líneas endogámicas S2
Se puede observar una tendencia en la distribución de las líneas en dos grupos principales.
La genética es un aspecto muy importante en la historia del maíz, con grandes poblaciones experimentales combinadas con genotipos de bajo costo, para favorecer la investigación rigurosa de sus mecanismos genéticos. Se han logrado grandes avances en los últimos años, pero aún quedan muchas preguntas por responder.
El costo de la genotipificación ha disminuido recientemente, por lo que es una herramienta útil para la selección asistida. Actualmente muchos investigadores se concentran en la correlación fenotipo-genotipo como las bases para la selección debido al beneficio económico que representan la reducción del costo y aceleramiento del proceso de selección. Los resultados de esta selección genómica son prometedores, pero aún existe mucho debate en el campo sobre si esta metodología es la mejor [20]; esto se confirma con los resultados obtenidos en esta investigación. De manera ideal, se puede considerar una comparación entre la selección genómica y la selección fenotípica estándar en un programa de mejoramiento, para evaluar su desempeño relativo y determinar qué tan bien se mantiene la precisión predictiva en la práctica. Los resultados obtenidos por otros investigadores [2] permiten asegurar que los SSR permiten estimar adecuadamente las relaciones y distancias genéticas entre grupos de organismos (plantas). Mediante esta técnica se lograron identificar 17 de 19 híbridos analizados y los genotipos más cercanos entre sí fueron aquellos que difirieron sólo en una característica, como resistencia a insectos o herbicidas [2]. Esto nos permite confiar en que la agrupación de nuestras líneas es justamente acorde a su grado de similitud; de hecho, las dos poblaciones (RR1 y RR2) derivadas de la población original se agrupan juntas, con casi 0.80 de similitud, lo que era de esperarse, no obstante que han sido sembradas y han sufrido selección posterior en distintos ambientes.
Acorde a lo esperado, los materiales aún muestran un alto grado de similitud, aunque su separación en grupos permitirá una mejor selección para los posibles futuros progenitores de híbridos potenciales, lo que permitiría también verificar los resultados aquí presentados con la genotipificacion futura de las líneas a medida que se avance en el nivel de endogamia [11].
En última instancia, la genética cuantitativa del maíz tiene como objetivo mejorar el rendimiento de maíz para la agricultura, de manera más eficiente respecto a la utilización de los recursos, lo que se puede lograr al aplicar la metodología empleada en el trabajo, con lo que se reducen costos y tiempos, impactando de manera positiva la economía de los productores y, por ende, de los consumidores.
Conclusiones
El análisis de la matriz de similitud demostró la semejanza entre las líneas 8 y 81, así como para RR1 Y RR2. Las líneas 81 y 8 poseen mayor variabilidad respecto a la población original, aunque la línea 81 tiene mayor similitud, 0.647, con la población original. El nivel de la endogamia dentro de las líneas es bajo, por lo que aún no se fijan los caracteres por los que se seleccionaron los materiales y las poblaciones no se agrupan por dichas cualidades. Pero, se requiere una cantidad mayor de marcadores para poder determinar una relación más clara entre las líneas endogámicas y la población original. Se generó información necesaria para poder continuar con el análisis de estas líneas y se sentaron las bases para continuar con el incremento de nivel de endogamia que permita fijar los caracteres deseados. A priori, las líneas más alejadas entre sí podrían usarse como progenitores del híbrido.
Agradecimiento
Al PRODEP por el apoyo del proyecto de “Fortalecimiento de Cuerpos Académicos” en favor del Cuerpo Académico “Biotecnología y Bioquímica de Semillas” del Instituto Tecnológico de Roque. Reporte de proyecto: PROMEP, IDCA: 9869. CLAVE: ITROQ-CA-2
Referencias
Lee, M. (1995). DNA markers and plant breeding programs. Adv. Agron ., 55, 265-344.
Bonamico, N., Aiassa, J., Ibañez, M., Di Renzo, M., Díaz, D., Salerno, J. (2004). Caracterización y clasificación de híbridos simples de maíz con marcadores SSR. RIA 33, 129-144.
Reyes-Castañeda, P. (1990). El maíz y su cultivo . Primera edición. AGT Editor. México. 640 pp.
SIAP (2012). Módulo Agrícola del SIACON 1980 - 2011. Servicio de Información Agroalimentaria y Pesquera. http://www.siap.gob.mx . Revisado el 20 de febrero de 2013. (Error 5: El enlace externo http://www.siap.gob.mx debe ser una url) (Error 6: La url http://www.siap.gob.mx no esta bien escrita)
Pérez-Taveras, R. A., Carballo-Carballo, A., Castillo-González, F. y Covarrubias-Prieto, J. (1991). Identificación de patrones heteróticos en un grupo de variedades precoces de maíz. Agrociencia serie Fitociencia 2, 69–79.
Gale, M. D. (1994). Genetic markers, maps and wheat breeding. J. R. Agríe. Soc. Engl. , 155, 162- 176.
Pérez de la Vega, M. (1993). Biochemical characterization of populations. ‘Plant Breeding: Principles and Prospects’. M. D. Hayward, N. O. Bosemark& l. Romagosa Eds. Chapman & Hall. London. pp 184-200.
Arús, P. y J. Moreno-González (1993). Markerassisted selection. En ‘Plant Breeding: Principies and Prospects’. M. D. Hayward, N. O. Bosemark& l. Romagosa Eds. Chapman & Hall. London. pp 314-331.
Stebbins, G. L. y M. Pérez de la Vega. (1989) Estimación de la variabilidad genética. En ‘Evolución hacia una Nueva Síntesis. Contribuciones desde el Reino Vegetal’ . G. L. Stebbins& M. Pérez de la Vega. Sec. Publ. Univ. de León, León. pp. 63-81.
Azofeifa-Delgado, A. (2006) Uso de marcadores moleculares en plantas: aplicaciones en frutales del trópico. Agronomía Mesoamericana . 17, 221-242.
Senior M. L., Murphy J. P., Goodaman M.M., and Stuber C. W. (1998). Utility of SSRs for determinating genetics similarities and relationships in maize using an agarose gel system. Crop Science 38, 1088-1098.
Schnable Patrick, S. et al (2009). The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 326, 112-115.
Gaut B. S., Le Thierry d’Ennequin M., Peek A. S., and Sawkins M. C. (2000). Maize as a model for the evolution of plant nuclear genome. Proceedings of the National Academy of the Sciences USA . 97, 7008-7015.
Tenaillon, M. I., Sawkins, M. C., Long, A. D., Gaut, R. L., Doebley, J. F. &Gaut, B. S. (2001) Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.). Proceedings of the National Academy of the Sciences of the USA 98, 9161–9166.
Jaenicke D. Viviane; Ed. Buckler; Bruce D Smith; M Thomas P Gilbert; Alan Cooper; John Doebley; Svante Pääbo. 2003. Early allelic selection in maize as revealed by ancient DNA. Science , 302, 1206-8.
Piperno, D.R., Anthony J. Ranere, Irene Holst, Jose Iriarte, and Ruth Dickau (2009). Starch grain and phytolith evidence for early ninthmillennium B.P. maize from the Central Balsas River Valley, Mexico. PNAS 106, 5019–5024.
Salinas-Jiménez, V., J. C. Raya-Pérez, C. L. Aguirre-Mancilla, F. Chablé-Moreno, J. G. Ramírez-Pimentel, G. García-Rodríguez, J. Covarrubias-Prieto (2015). Prueba temprana en líneas S1 de maíz. En prensa.
Liu, Y. G.,Mitsukawa N., Oosumi T., and Whittier R. F. (1995). Efficient isolation and mapping of Arabidopsis thaliana T-DNA insert junctions by thermal asymmetric interlaced PCR. Plant Journal 8, 457-463.
Rohlf, F. J. (1998). NTSYS–PC, Numerical taxonomy and multivariate analysis system. Version 1.60. Applied Biostatistics, New York.
Heslot N, Yang H-P, Sorrells ME, Jannink J-L (2012). Genomic selection in plant breeding: a comparison of models. Crop Sci , 52, 146–160.
Notas de autor
ceaguirre@itroque.edu.mx