Caso clínico
Received: 12 December 2023
Accepted: 15 August 2024
DOI: https://doi.org/10.29262/ram.v71i4.1399
Resumen
Antecedentes: El síndrome de deleción 22q11 consiste en una agrupación variable de características fenotípicas y defectos inmunológicos, secundarias a la pérdida del material genético localizado en la banda 22q11.2. El espectro de deleción del 22q11 abarca diferentes síndromes relacionados con la misma etiología y con anomalías superpuestas, incluidos el síndrome de DiGeorge, síndrome velocardiofacial, entre otros.
Reporte de caso: Paciente masculino de 6 meses de edad, de etnia indígena Rarámuri, enviado al hospital Infantil de Especialidades de Chihuahua por desnutrición severa y dificultad respiratoria. A su ingreso se detectó un soplo holosistólico grado V/VI y retraso global del neurodesarrollo. Requirió intubación endotraqueal e ingresó a la Unidad de terapia intensiva pediátrica, donde tuvo una larga estancia hospitalaria. Se sospechó algún tipo de error innato de inmunidad por procesos infecciosos, cardiopatía congénita y datos de dismorfias faciales (hipoplasia malar, hendiduras palpebrales estrechas, nariz aspecto tubular y boca pequeña con paladar ojival), además de síndrome de DiGeorge.
Conclusiones: El síndrome por deleción 22q11 provoca manifestaciones clínicas heterogéneas, incluidas múltiples anormalidades cardiacas y enfermedades crónicas. En la población Rarámuri, actualmente no existen reportes de este tipo de errores innatos de la inmunidad, por lo que nuestro paciente ejemplifica la necesidad de buscar intencionadamente estos trastornos, sobre todo en quienes padecen infecciones severas, cardiopatías y características morfológicas distintivas. Es necesario llevar a cabo programas de detección de enfermedades, particularmente en poblaciones vulnerables.
Palabras clave: Síndrome de deleción 22q11, Síndrome de DiGeorge, Errores innatos de la inmunidad, Población Rarámuri, Chihuahua.
Abstract
Background: 22q11 deletion syndrome consists of a variable grouping of phenotypic features and immunological defects secondary to the loss of genetic material located in the 22q11.2 band. The 22q11 deletion spectrum encompasses different syndromes related to the same etiology and with overlapping anomalies, including DiGeorge syndrome, velocardiofacial syndrome, among others.
Case report: A 6-month-old male patient of indigenous Rarámuri ethnicity was referred to the Children's Specialty Hospital of Chihuahua due to severe malnutrition and respiratory distress. Upon admission, a grade V/VI holosystolic murmur and global neurodevelopmental delay were detected. He required endotracheal intubation and was admitted to the Pediatric Intensive Care Unit, where he had a long hospital stay. Some type of inborn error of immunity was suspected due to infectious processes, congenital heart disease, and evidence of facial dysmorphisms (malar hypoplasia, narrow palpebral fissures, tubular-like nose, and small mouth with high-arched palate), in addition to DiGeorge syndrome.
Conclusions: 22q11 deletion syndrome causes heterogeneous clinical manifestations, including multiple cardiac abnormalities and chronic diseases. In the Rarámuri population, there are currently no reports of this type of inborn error of immunity, so our patient exemplifies the need to intentionally search for these disorders, especially in those with severe infections, heart disease, and distinctive morphological characteristics. Disease detection programs are necessary, particularly in vulnerable populations.
Keywords: 22q11 deletion syndrome, DiGeorge syndrome, Inborn errors of immunity, Rarámuri population, Chihuahua.
ANTECEDENTES
El síndrome de DiGeorge es un trastorno que afecta frecuentemente al cromosoma 22, ocasionando la deleción 22q11. El síndrome de DiGeorge es la microdeleción más común y suele subdiagnosticarse en diversas poblaciones. La prevalencia estimada varía de 1:3000 a 1:6000 niños. 1 El síndrome de DiGeorge se caracteriza por enfermedad cardiaca congénita (defectos conotruncales), inmunodeficiencia, hipoparatiroidismo, alteraciones gastrointestinales, esqueléticas y renales, con características faciales distintivas. 2 La mayor parte de los estudios se han enfocado en individuos de ascendencia europea; sin embargo, algunos reportes demuestran que en ciertas poblaciones, por ejemplo China, o en sujetos afrodescendientes, el defecto es subdiagnosticado.
A continuación se expone un caso representativo en la población Rarámuri de Chihuahua, México.
REPORTE DE CASO
Paciente masculino de 6 meses de edad, de etnia indígena Rarámuri, originario de Urique, Chihuahua, nacido de un segundo embarazo (normoevolutivo), mediante parto, con peso de 2650 g, control prenatal regular, madre de 25 años, sana; y padre finado, de quien se desconoce la causa. Vivienda rural, en hacinamiento, sin servicios básicos; vacunación incompleta. Fue enviado al hospital Infantil de Especialidades de Chihuahua por desnutrición severa y dificultad respiratoria. A su ingreso se detectó un soplo holosistólico grado V/VI y retraso global del neurodesarrollo. Requirió intubación endotraqueal e ingresó a la Unidad de terapia intensiva pediátrica, donde tuvo una larga estancia hospitalaria, periodo en el que manifestó tres eventos de neumonía, con aislamiento de Staphylococcus aureus en una ocasión y urocultivo positivo para Candida albicalns. Padeció neumopatía crónica, con datos de aspiración y alteración en la mecánica de la deglución. Durante su estancia hospitalaria requirió múltiples esquemas de antibióticos, apoyo aminérgico y transfusión de hemoderivados. La valoración por personal del servicio de Cardiología detectó cardiopatía congénita tipo CIV amplia de 8 mm, con hipertensión arterial pulmonar severa (68 mmHg), CIA tipo foramen oval permeable y PCA.
Se sospechó algún tipo de error innato de inmunidad por procesos infecciosos, cardiopatía congénita y datos de dismorfias faciales (hipoplasia malar, hendiduras palpebrales estrechas, nariz, aspecto tubular y boca pequeña con paladar ojival; Figura 1Ay1B), además de síndrome de DiGeorge. El 5/02/2021 se llevó a cabo el estudio de fluorescencia con hibridación in situ (FISH), que reportó deleción de la secuencia específica LSI TUPLE1 (22q11.25).
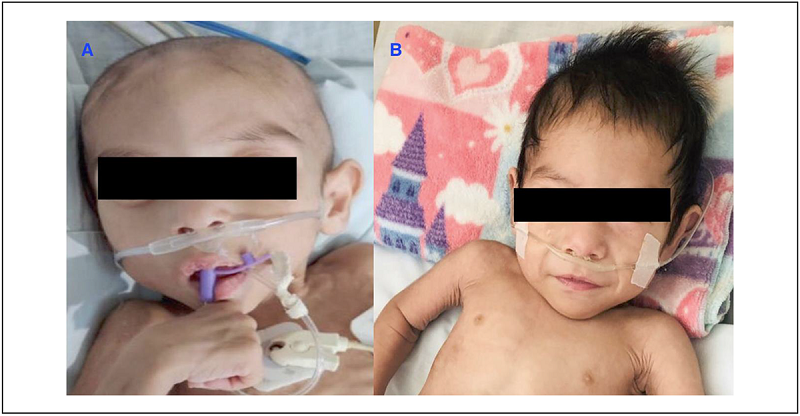
Figura 1.
A) Paciente con ocho meses de edad. Se observa discreta hipoplasia malar, hendiduras palpebrales estrechas, nariz con aspecto tubular y boca pequeña con paladar ojival. B) Paciente a los 4 años de edad.
Los estudios de laboratorio informaron: panel de inmunoglobulinas (08/01/2021): IgA 72 mg/dL, IgG 436 mg/dL, IgM 59.7 mg/dL, IgE 477 UI/mL. El 09/02/2021 se detectaron subpoblaciones linfocitarias con linfopenia (1792 linfocitos totales), linfocitos T CD3+(1220/μL) CD4+(876/μL) CD8 (317/μL), CD19+ (304//μL), y CD16+CD56+ (211/μL). La radiografía de tórax de ingreso con pobre sombra tímica Figura 2A. El 24/04/2020 se solicitó una tomografía computada de tórax, que reportó hipoplasia tímica. Figura 2B
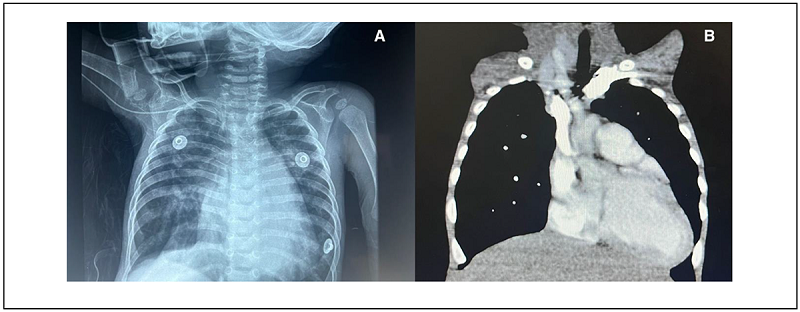
Figura 2.
A) Radiografía de tórax al ingreso hospitalario. B) Tomografía de tórax que muestra hipoplasia tímica.
Debido a la desnutrición severa e infecciones de repetición, se inició tratamiento con gammaglobulina intravenosa, en dosis de 1 g/kg mensual, con lo que tuvo reacción satisfactoria de los procesos infecciosos. Se realizó cerclaje pulmonar y posterior cierre de CIV y CIA con circulación extracorpórea. La evolución fue favorable, con mejoría del estado nutricional y control de los procesos infecciosos. Se dio de alta por mejoría clínica; sin embargo, hubo pérdida de seguimiento por más de tres años.
Reingresó al hospital a los 4 años de edad, con neumonía severa, descompensación de la cardiopatía y, nuevamente, desnutrición severa (peso de 10.8 kg y talla de 89 cm). La tomografía de tórax reportó consolidación posterior en ambos campos pulmonares, con patrón de vidrio despulido y atelectasias laminares, ameritando su estancia en la Unidad de terapia intensiva de Pediatría, soporte ventilatorio fase III y amplio esquema antimicrobiano, con evolución satisfactoria. Actualmente, se encuentra estable. Se reinició el tratamiento con inmunoglobulina intravenosa mensual.
DISCUSIÓN
La descripción del epónimo del síndrome de DiGeorge, establecido por Angelo DiGeorge (1965), incluyó infantes con ausencia de timo (aplasia tímica) y glándulas paratiroides (hipoparatiroidismo). 1 Las alteraciones cardiacas congénitas, especialmente las que involucran el conducto de salida, se añadieron posteriormente a la lista de síntomas, contribuyendo a la teoría de la perturbación del desarrollo embrionario del tercer y cuarto arco faríngeo. 2 4 Hoy día se conoce que el síndrome provoca manifestaciones clínicas heterogéneas, por ejemplo: alteraciones gastrointestinales, renales, autoinmunidad, trastornos psiquiátricos y distintos grados de inmunodeficiencia. 3 En el caso aquí expuesto, la sospecha diagnóstica basada en infecciones severas, cardiopatía y hallazgos morfológicos distintivos llevó a la búsqueda intencionada de errores innatos de la inmunidad con características sindrómicas.
El síndrome de deleción 22q11.2 es el desorden cromosómico por microdeleción más frecuente en pacientes con síndrome de DiGeorge.
La hipoplasia tímica da como resultado rango variable de disfunción de células T. La mayoría de los pacientes con síndrome de DiGeorge tiene defectos leves en la cantidad de células T y, clínicamente, carecen de un comportamiento de inmunodeficiencia severa. Sin embargo, puede haber un espectro en el grado de linfopenia, y del 0.5 al 1% de los casos tienen completa ausencia de timo, lo que resulta en una severa inmunodeficiencia. 3 5 La atimia congénita se conoce como síndrome de DiGeorge completo y se reconoce por concentraciones menores de 5% de linfocitos T vírgenes/mm 3 o menos del 5% del total de linfocitos-T, que sugiere un tipo de inmunodeficiencia combinada grave y amerita trasplante de timo. Una forma igualmente grave, conocida como forma atípica de síndrome de DiGeorge, se caracteriza por exantema y linfocitos T oligoclonales circulantes. 4
En la paciente de este caso, quien manifestó hipoplasia tímica, se observó un cuadro clínico de inmunodeficiencia, con infecciones pulmonares severas y diferentes grados de linfopenia, por lo que el mantenimiento de terapia con inmunoglobulina endovenosa ha permitido una mejor respuesta a las infecciones. En este sentido se ha documentado un déficit funcional de linfocitos-B, ocasionalmente con hipogammaglobulinemia, en pacientes con síndrome de DiGeorge. En un estudio internacional se encontró que 6% de los pacientes mayores de 3 años tuvieron hipogammaglobulinemia y 3% de esta cohorte de 1023 pacientes con síndrome de DiGeorge se encontraron con gammaglobulina sustitutiva. Además, se ha encontrado que disminución progresiva de linfocitos-B de memoria por afectación de cooperación con el linfocito-T. 6
Los informes de pacientes latinoamericanos con síndrome de DiGeorge son particularmente escasos. En México, Márquez-Ávila 8 reportó 62 pacientes con síndrome de deleción 22q11.2, y de estos 37% tuvieron infecciones recurrentes. 7 Ramírez-Velazco emprendió un estudio con 39 pacientes con dismorfias sugerentes de con síndrome de DiGeorge, y de estos 22 tuvieron deleción 22q11.2 y 14% inmunodeficiencia; sin embargo, no existen reportes de este tipo de estudios. 8
En cuanto a la población Rarámuri, se carece de información acerca de errores innatos de la inmunidad. Del Rio y Nájera y colaboradores 9 realizaron un estudio para determinar la prevalencia de dolor musculoesquelético y enfermedad reumatológica en la población Rarámuri mayor de 18 años. Informaron una prevalencia de enfermedad reumática del 10.5%, con diagnósticos específicos de osteoartritis (6.6%), dolor lumbar (1.6%), espondiloartritis (0.8%), artritis reumatoide (0.5%), síndrome de dolor articular (0.5%) y fibromialgia (0.4%). 9
El caso aquí reportado supone el primero informado en la población Tarahumara, con hallazgos faciales característicos de síndrome de DiGeorge. Puesto que existen pocos informes de enfermedad inmunológica en la población Rarámuris, consideramos que en los países en vías de industrialización y particularmente en ciertas etnias, el diagnóstico puede estar infravalorado.
CONCLUSIÓN
El síndrome por deleción 22q11 provoca manifestaciones clínicas heterogéneas, incluidas múltiples anormalidades cardiacas y enfermedades crónicas. Nuestro paciente tuvo la triada de cardiopatía, hipoplasia tímica e infecciones severas, lo que refleja la importancia del estado de inmunodeficiencia, que suele ser variable en esta patología y, por lo tanto, es necesario individualizar el tratamiento, particularmente la aplicación de gammaglobulina endovenosa en pacientes con la forma completa, incluso incompleta, de síndrome de DiGeorge.
En la población Rarámuri, actualmente no existen reportes de este tipo de errores innatos de la inmunidad, por lo que nuestro paciente ejemplifica la necesidad de buscar intencionadamente estos trastornos, sobre todo en quienes padecen infecciones severas, cardiopatías y características morfológicas distintivas. Es necesario llevar a cabo programas de detección de enfermedades, particularmente en poblaciones vulnerables.
REFERENCIAS
Sullivan KE. Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial syndrome. Immunol Allergy Clin North Am. 2008; 28 (2): 353-66.
Kuo CY, Signer R, Saitta SC. Immune and Genetic Features of the Chromosome 22q11.2 Deletion (DiGeorge Syndrome). Curr Allergy Asthma Rep. 2018; 18 (12): 75.
Sullivan KE. DiGeorge syndrome and chromosome 22q11.2 deletion syndrome. In: Ochs HD, Stiehm ER, Winkelstein JA, eds. Immunologic Disorders in Infants and Children. 5th ed. Philadelphia: Elsevier; 2004: 523.
Biggs SE, Gilchrist B, May KR. Chromosome 22q11.2 Deletion (DiGeorge Syndrome): Immunologic Features, Diagnosis, and Management. Curr Allergy Asthma Rep. 2023; 23 (4): 213-222.
Derfalvi B, Maurer K, McDonald-McGinn DM, et al. B cell development in chromosome 22q11.2 deletion syndrome. Clin Immunol. 2016; 163: 1-9.
Addissie YA, McGinn DE, Porras AR, et al. 22q11.2 deletion syndrome in diverse populations. Am J Med Genet A. 2017; 173 (4): 879-888.
Márquez-Ávila CS, Vizcaíno-Alarcón A, García-Delgado C, et al. Velocardiofacial syndrome in Mexican patients: Unusually high prevalence of congenital heart disease. Int J Pediatr Otorhinolaryngol. 2015; 79 (11): 1886-1891.
Ramírez-Velazco A, Rivera H, Vásquez-Velázquez AI, et al. 22q11.2 deletion detected by in situ hybridization in Mexican patients with velocardiofacial syndrome-like features. Colomb Med (Cali). 2018; 49 (3): 219-222.
Del Río Nájera D, Santana N, Peláez-Ballestas I, et al. Prevalence of rheumatic diseases in Raramuri people in Chihuahua, Mexico: a community-based study. Clin Rheumatol. 2016; 35 (Suppl 1): 43-52.
Financiamiento
Author notes
*Correspondencia: Luisa Berenise Gámez González. dra.luisa.gamez@gmail.com
Conflict of interest declaration
Los autores declaran no tener conflicto de interés.
